
30.2: Pinacol Rearrangement
- Page ID
- 44001
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The pinacol rearrangement was the first molecular rearrangement identified as such by early chemists. The defining example of a pinacol rearrangement is shown in the following diagram. Pinacol itself is produced by magnesium reduction of acetone, probably by way of a ketyl intermediate. Since the diol is symmetrical, protonation and loss of water takes place with equal probability at either hydroxyl group. The resulting 3º-carbocation is relatively stable, and has been shown to return to pinacol by reaction in the presence of isotopically labeled water. A 1,2-methyl shift generates an even more stable carbocation in which the charge is delocalized by heteroatom resonance. Indeed, this new cation is simply the conjugate acid of the ketone pinacolone, which is the product of repeated rearrangements catalyzed by proton transfer. Each step in this rearrangement is potentially reversible, as demonstrated by the acid catalyzed dehydration of pinacolone (and pinacol) to 2,3-dimethyl-1,3-butadiene under vigorous conditions.
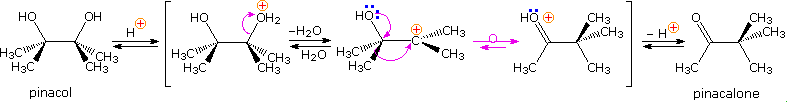
Many factors must be considered when analyzing the course of a pinacol rearrangement. These include:
• Which hydroxyl group is lost as water? or Which intermediate carbocation is more stable?
• What is the inherent shifting tendency (migratory aptitude) of different substituent groups?
• What is the influence of steric hindrance and other strain factors on the rearrangement?
• Are epoxides formed as intermediates in the pinacol rearrangement?
• Does product stability govern the outcome of competing rearrangements?
• Do the reaction conditions (i.e. type of acid, concentration, solvent and temperature) influence the course of rearrangement?
Virtually all of these factors have been shown to be important in one or more cases, and a full analysis of their complex interaction is beyond the scope of this text. Nevertheless, a few examples will be presented to demonstrate the general nature of this transformation, and to illustrate the action of some of the above factors. In the first reaction shown below, we see an example of kinetic versus thermodynamic product control. Under mild acid treatment, the diol rearranges rapidly to an aldehyde by way of a 1,2-hydrogen shift to the initially formed diphenyl 3º-carbocation. More vigorous acid treatment of the diol or the aldehyde generates the more stable phenyl ketone (conjugation of the phenyl and carbonyl groups). Mechanisms for this and the other reactions will be presented by clicking on the diagram. A pink colored arrow designates rearrangement; light blue arrows indicate epoxide ring closing or opening reactions. Repeated clicking toggles the reaction and mechanism displays.
The second example describes a similar reacting system, which provides additional information from stereochemical and isotopic labeling features. Loss of water from the 3º-carbinol site, followed by a reversible 1,2-hydride shift, generates the conjugate acid of the ketone product. At short reaction times, racemization of recovered diol starting material occurs at the same rate as rearrangement. A corresponding phenyl shift to the initially formed 3º-carbocation generates the aldehyde conjugate acid, and the aldehyde itself has been shown to isomerize to the same rearranged ketone under the conditions of this pinacol rearrangement. An isotopic carbon label (colored green) in either the diol or aldehyde is scrambled (colored brown) in the course of these reactions, suggesting an epoxide intermediate.
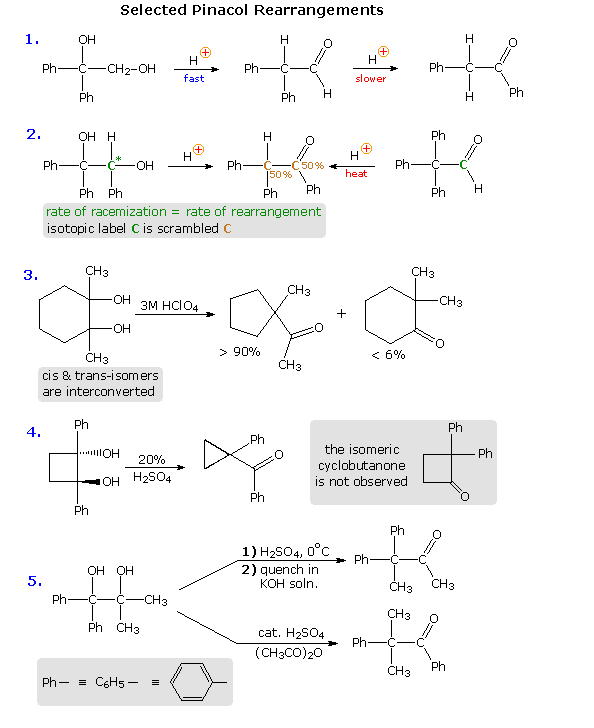
In reaction # 3 either the cis or trans diol may be used as a reactant. These isomers are rapidly interconverted under the rearrangement conditions, indicating that the initial water loss is reversible; a result confirmed by isotopic oxygen exchange. The clear preference for a methylene group shift versus a methyl group shift may reflect inherent migratory aptitudes, or possibly group configurations in the 3º-carbocation intermediate. In the conformation shown here both methyl and methylene groups may shift, or an epoxide ring may be formed reversibly. An alternative chair-like conformation having an equatorial methyl group should be more stable, but would not be suitable for a methyl shift. The predominant ring contraction is therefore understandable. Reaction # 4 is an unusual case in which a strained ring contracts to an even smaller ring. Phenyl groups generally have a high migratory aptitude, so the failure to obtain 2,2-diphenylcyclobutanone as a product might seem surprising. However, the carbocation resulting from a phenyl shift would be just as strained as its precursor; whereas the shift of a ring methylene group generates an unstrained cation stabilized by phenyl and oxygen substituents. Conjugative stabilization of the phenyl ketone and absence of sp2 hybridized carbon atoms in the small ring may also contribute to the stability of the observed product.
Finally, reaction # 5 clearly shows the influence of reaction conditions on product composition, but explaining the manner in which different conditions perturb the outcome is challenging. Treatment with cold sulfuric acid should produce the more stable diphenyl 3º-carbocation, and a methyl group shift would then lead to the observed product. The action of a Lewis acid in acetic anhydride, on the other hand, may selectively acetylate the less hindered dimethyl carbinol. In this event the acetate becomes the favored leaving group (presumably coordinated with acid), followed by a 1,2-phenyl shift. It is reported that the symmetrically substituted isomeric diol (drawn in the shaded box) rearranges exclusively by a methyl shift, but the configuration of the starting material was not stated (two diastereomers are possible).
Because of the influence of other factors (above), it has not been possible to determine an unambiguous migratory order for substituents in the pinacol rearrangement. However, some general trends are discernible. Benzopinacol, (C6H5)2C(OH)C(OH)(C6H5)2, undergoes rapid rearrangement to (C6H5)3CCOC6H5 under much milder conditions than required for pinacol. Indeed, it is often the case that phenyl or other aromatic substituents adjacent to a forming carbocation will facilitate that ionization in the course of their migration to the cationic site. In non-aromatic compounds of the type (CH3)2C(OH)C(OH)RCH3, migration of R increases in the manner R = CH3 < R = C2H5 << R = (CH3)3C. Since the shifting alkyl group must carry part of the overall positive charge, alkyl substitution should have a stabilizing influence on the rearrangement transition state. Finally, fluorine substitution, as in C6H5(CF3)C(OH)C(OH)(CF3)C6H5 renders the diol unreactive under acid catalyzed rearrangement conditions. Here, the powerful inductive withdrawal of electrons by fluorine inhibits positive charge formation.