2.5E: GC Parameters
- Page ID
- 95703

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)There are many factors that affect the separation (and/or retention times) in gas chromatography, including the type of column, sample concentration, oven temperature, and flow rate of carrier gas. The factors controllable by a student are described in this section, and are related to concentration and temperature.
Dilution
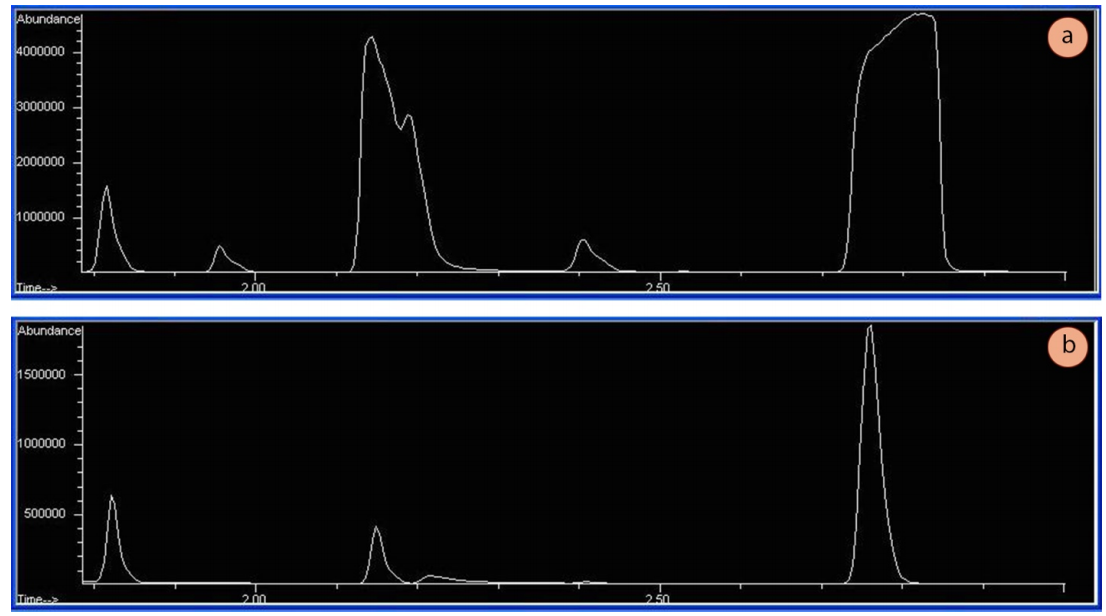
GC runs must be conducted on very dilute samples. A dilute sample enables adequate stationary-mobile phase equilibrium inside the column, resulting in narrow Gaussian-shaped peaks. If samples are too concentrated, the peak shapes will often be broad and flattened on top (Figure 2.87a), indicating the column (and/or detector) has been overwhelmed. Appropriate abundances (the y-axis on GC spectra) using a mass-spectrometer detector should be in the low millions (e.g. 1,500,000 TIC's or so, see Figure 2.87b).
A GC should never be run on undiluted liquid. Overly concentrated samples do not only cause peaks to broaden and possibly overlap, they can also be harmful to detectors. Mass spectrometers require very small quantities of liquid, or over time, their filaments degrade.
To prepare a routine GC sample, \(1.5 \: \text{mL}\) of a low-boiling solvent (e.g. methanol, clean acetone, diethyl ether, or dichloromethane) is added to a GC vial (Figure 2.88) along with one drop of compound. the instrument then injects roughly \(1 \: \mu \text{L}\) of this dilute solution into the instrument, which is further diverted internally, resulting in very small quantities entering the narrow capillary column and detector. The normal function of a GC instrument requires tiny quantities of material, which is a major advantage to this technique.

Solvent Delay (With Mass Spectrometers)
A properly prepared GC sample contains a small amount of compound dissolved in a solvent, and yet GC spectra (when paired with a mass spectrometer detector) often do not show any solvent peaks. The reason is that GC methods often include a "solvent delay", where the detector is turned off until a certain amount of time has passed after injection. Even with the dilution inside the GC-MS instrument, the quantity of solvent reaching the detector would overwhelm and eventually degrade it. Therefore, the detector is left inactive until the solvent has passed through the column, and activated afterwards.
If you look at any GC-MS spectrum, the retention time (x-axis) never starts at zero minutes, which represents the time the sample is injected on the column. In the GC spectrum of hexanes, the spectrum starts at 1.40 minutes, (as is indicated with an arrow in Figure 2.89). The solvent would have eluted from the column before this time.
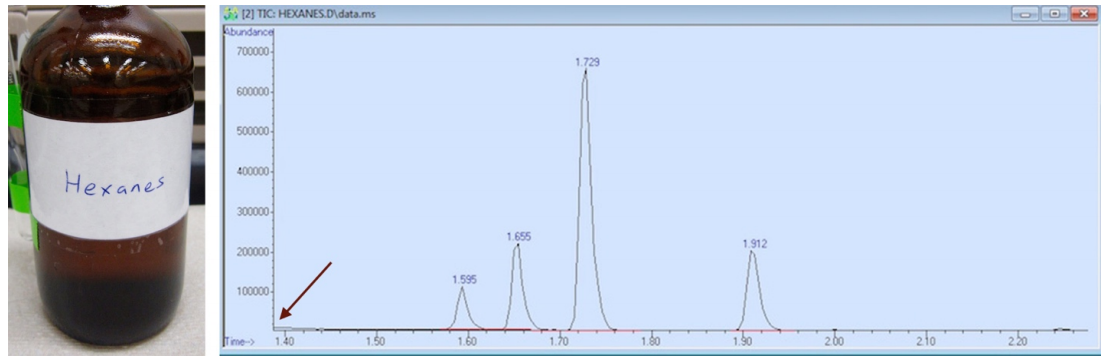
The GC solvent must be chosen such that the solvent will elute before the compound of interest (the solvent must have a significantly lower boiling point than the compound), so that the solvent delay may be set between elution of the solvent and sample. For example, methanol (b.p. \(65^\text{o} \text{C}\)) would have been an unsuitable solvent for the sample of hexanes (b.p. \(68^\text{o} \text{C}\)). If the solvent delay was set such that the hexane signals could be detected, the GC spectrum would have been overwhelmed by signals from the methanol, as the two compounds have very similar boiling points and would elute rather closely. If the detector was activated after the methanol had eluted, the instrument would likely miss detection of the closely eluting hexanes. Therefore, a significantly lower boiling solvent was used (diethyl ether, b.p. \(35^\text{o} \text{C}\)) to produce the GC spectrum of hexanes in Figure 2.89, and this solvent would have eluted before the solvent delay of 1.40 minutes.
Oven Temperature
The temperature of the GC oven is an easily adjusted variable (although it should be adjusted by an instructor, not student). The oven temperature has a dramatic effect on retention times, much in a similar way that changing the mobile phase in TLC has on \(R_f\).
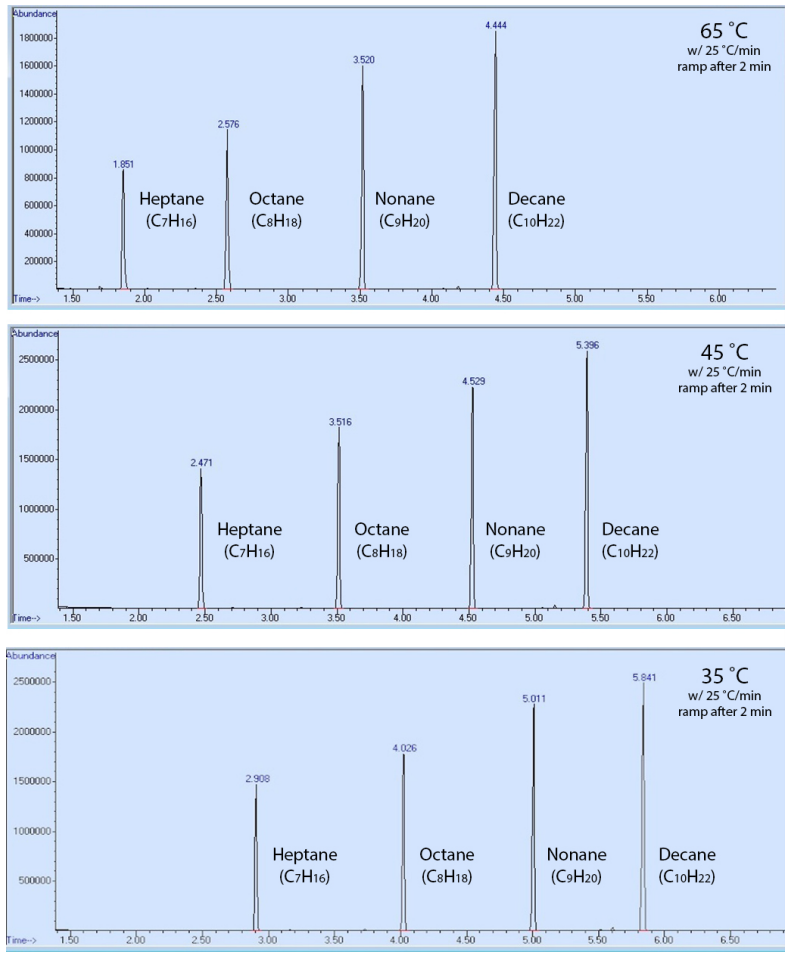
To demonstrate, a sample containing heptane, octane, nonane, and decane was run using three GC methods, differing only in their starting temperature (\(65^\text{o} \text{C}\), \(45^\text{o} \text{C}\), or \(35^\text{o} \text{C}\)). The order of elution of the compounds was the same in all methods (Figure 2.90), but the components eluted faster with a hotter oven temperature (had lower retention times). This result can be explained in that the compounds were more able to overcome their intermolecular attraction with the stationary phase (the coating of the GC column) when there was more available thermal energy. This resulted in compounds spending less time adhering to the stationary phase, and less time retained by the column.
Using a Temperature Ramp
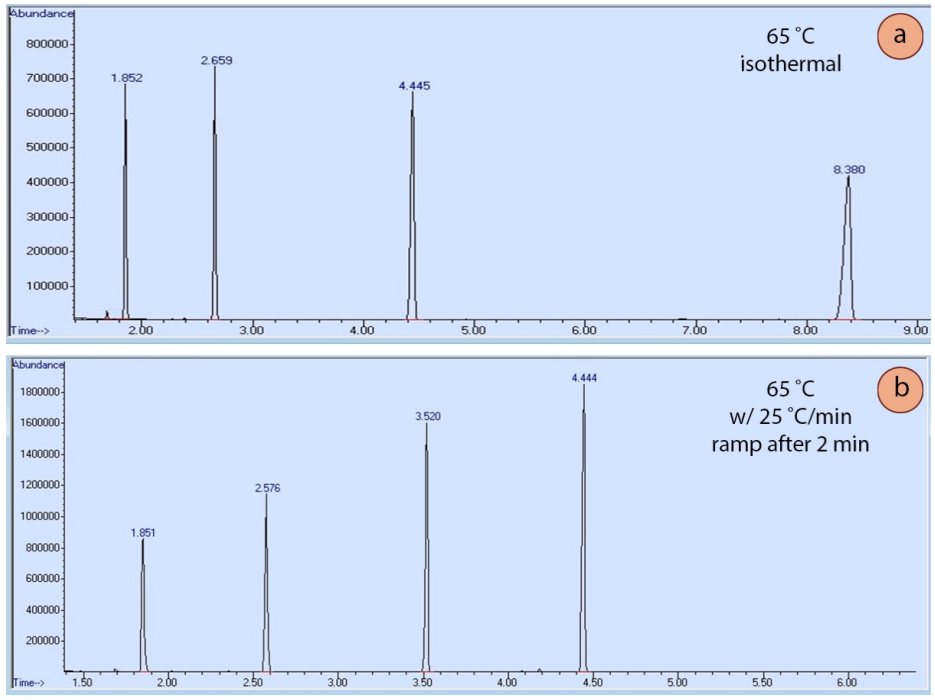
A GC can be run at one oven temperature the entire time (an "isothermal" run), or the method may include temperature programming. In a programmed method, the temperature may remain fixed for a certain length of time, and then may include a "ramp", where the temperature is increased at a steady rate during the run. A graphical display of a ramp generated by the GC instrument is shown in Figure 2.91.
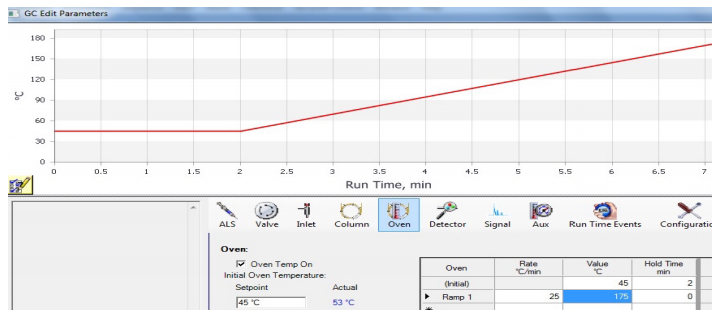
Temperature programming is useful because the longer a compound stays in the column, the wider it elutes as diffusion broadens the sample (analogously to how spots broaden during elution in TLC). Figure 2.92a shows an isothermal GC run of four compounds, and the peak shape significantly broadens as the retention time increases. Figure 2.92b shows a GC run of the same four compounds eluted using a temperature ramp. The ramp allows the compounds to elute faster, and for peak shape to sharpen and improve. The optimal ramp for each situation often takes experimentation to achieve the best peak shape and separation of components.