
1.31: Electrophilic Substitution
- Page ID
- 81797

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Electrophiles and Products
Let's begin by recalling the key steps in an electrophilic aromatic substitution mechanism.
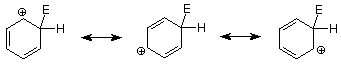
An important feature of this mechanism is that we can identify the electrophile if we know the product because it is the atom or group which replaces the H+. Conversely, if we know the electrophile, we can predict the structure of the product. The following table outlines these relationships in more detail for several reactions which follow the electrophilic aromatic substitution pathway.
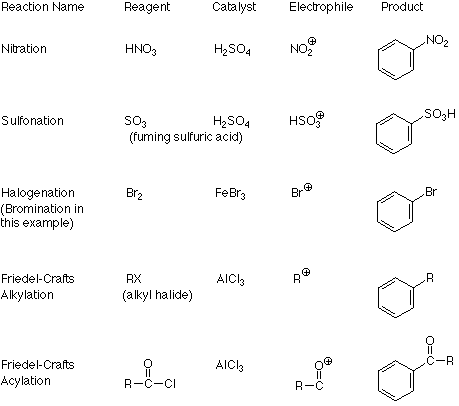
The connection between the electrophile and the product is clear from these examples. What is not clear is how the catalyst transforms the reagent into the electrophile. There are two patterns here. One applies when the electrophile is made by removing a halide ion from the reagent (halogenation and the two Friedel-Crafts reactions). The formation of a carbocation from an alkyl halide and aluminum trichloride is a typical example of the first process:
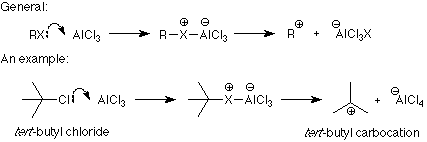
Notice that the role of the catalyst is to bond with the leaving group and make it into a better leaving group. This makes the formation of the electrophile (carbocation in this case) much easier so that there is more electrophile around to attack the benzene ring. Similar mechanisms are used in the halogenation and Friedel-Crafts acylation reactions.
For sulfonation and nitration, the catalyst is sulfuric acid. Just as was the case in the SN1 and SN2 reactions of alcohols, the function of the acid is to protonate an unshared electron pair on oxygen. When the OH group of nitric acid is involved, the OH2+group which is formed is a good leaving group. Its departure makes the active electrophile:
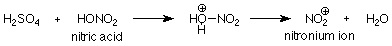
To summarize, the function of the catalyst is to convert the reagent into a strong electrophile, which then attacks the pi electrons on the aromatic ring to make a new covalent bond. The reaction is completed by the loss of an H+.
Ortho-Para Directive Effects
The reagent - catalyst - electrophile - product pattern works well when the aromatic compound is benzene itself, but things become more complicated when a substituted benzene (a molecule in which one of the hydrogens of benzene has been replaced by another atom or group) is used. There are three products which can arise in such a system. As an example let's look at toluene -- which is methyl benzene. We'll use nitration as our example of electrophilic aromatic substitution here.
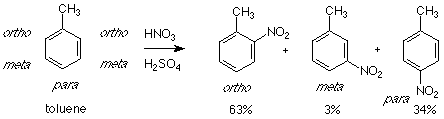
Although there are five hydrogens available to be replaced on the benzene ring of toluene , two of those are directly adjacent (ortho) to the methyl group. Attack at either of these positions gives the same product. Similar considerations apply to the meta position. The result is that only three products are obtained. The actual distribution of these products shows that very little of the meta product is obtained. Instead almost all of the product consists of the ortho and para isomers. For this reason the methyl group is called an "ortho-para directing group." It "directs" the incoming electrophile to attack at the ortho and para positions relative to itself.
How does the methyl group exert this "directive" effect? We can begin to make sense of this by remembering that we explained Markovnikov's rule by considering that the product which is most abundant is formed fastest through a pathway which has the lowest activation energy. That lead us to a consideration of the relative energies of competing transition states and the use of intermediates as useful "stand-ins" for those transition states. We concluded that Markovnikov's rule is explained by the idea that the more abundant products are formed by pathways which go through lower energy intermediates.
If we apply the same reasoning to these directive effects, the question becomes "What makes the intermediates for the formation of ortho and para products more stable than those for para products? As usual, we will look for the answers by comparing the structures of the intermediates.
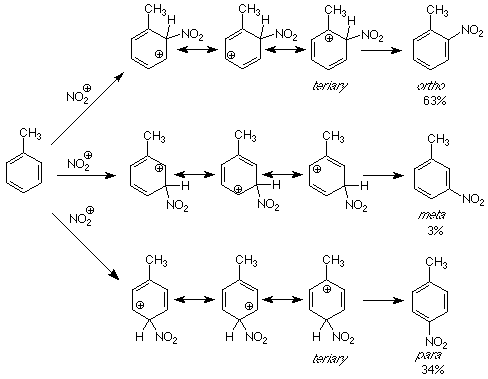
Each of the intermediates is described by resonance hybrid which includes three carbocations. However, for the intermediates leading to the ortho and para products, one of the contributing carbocationic structures is tertiary. This structure is more stable than the others because the electrons on the methyl group can directly stabilize the electron deficient carbocationic carbon. This stability is passed on to the resonance hybrid, which makes the intermediates for attack at the more stable than that for attack at the meta position. More stable intermediates mean lower energy transition states and faster reactions. A faster reaction means more product is formed through that pathway.
The result of this analysis is that groups which can donate electrons stabilize carbocations. Electrophilic attack will be faster at positions such that the carbocations produced have positive charges on carbons which are bonded to electron donating groups like methy groups. This occurs when attack happens at positions ortho and para to the electron donating group. Such electron donation happens with alkyl groups in general and when atoms like oxygen and amino-nitrogen are directly attacked to the ring. In the latter cases it is the unshared pair on the oxygen or nitrogen which helps reduce the energy of the electron deficient carbon atom. (See the figure at the bottom of p 165 in Atkins & Carey for an example.) Examine the ortho-para directing groups in Table 6.3 (p 162 of Atkins & Carey) and decide how each one acts as an electron donating group.
Meta Directive Effects
Can we extend this analysis to groups which are meta directing? Let's try an example. In the nitration of benzaldehyde the product mixture is 19% ortho, 72% meta, and 9% para. The aldehyde carbonyl group (and carbonyl groups in general) is a metadirecting substituent. We'll look at the resonance structures of the intermediate for an explanation:
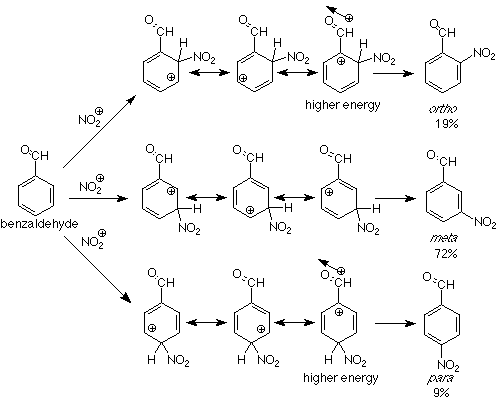
We see that attack at positions ortho and para to the carbonyl group places the positive charge directly adjacent to the positive end of the carbonyl group's dipole. The energy of such an intermediate will be higher than one in which the positive charge never approaches the carbonyl carbon, which is the case in the intermediate for meta attack. The consequence is that attack at the meta position is faster and more meta product is formed. Groups in which the atom directly attached to the benzene ring have a partial or complete positive charge tend to pull electrons toward themselves. Such groups are called electron withdrawing groups and are meta directing groups in electrophilic aromatic substitution. Look at the meta directing groups in Table 6.3 (p 162 in Atkins & Carey) and see if the preceding statement is borne out.
Activation and Deactivation
The underlying idea in the forgoing analysis of directive effects in electrophilic was understanding the relative rates of reaction when the competition was between different sites in the same molecule. This is termed intramolecular competition and it was also used in our understanding of Markovnikov's rule. We can also look at competition between molecules -- intermolecular competition -- using the same ideas, comparing the energies of intermediates in competing pathways. For example, let's imagine a reaction in which we mix toluene and benzene and then do a nitration. If we get more nitrotoluene (all three isomers) than nitrobenzene, the toluene has reacted faster than the benzene. We say that the methyl group "activated" the benzene ring. We understand this to mean that the methyl group lowered the energy of intermediates and transition states along the pathway to product as compared to the effect of a hydrogen (in benzene).
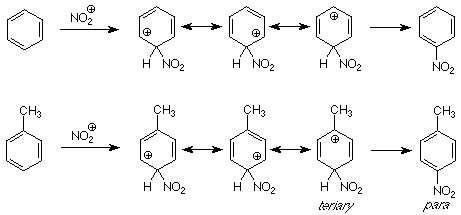
A comparision of the intermediates along the pathways from benzene and toluene to their respective products shows us the reason. (Only the para pathway is shown; the ortho pathway is similar.) The intermediate for attack para to the methyl group includes one resonance structure which is tertiary and thus is stabilized by the electron donating character of the methyl group. There is no such structure in the intermediate for nitration of benzene, so the intermediate for toluene nitration is more stable and the reaction which goes through it is faster. We call the methy group (and alkyl groups in general) an "activating" group for electrophilic aromatic substitution.
Since the electron donating characteristics of the methyl group are responsible for both its activating properties and its ortho-para directing properties, we might suspect that ortho-para directing groups will also be activating groups. An examination of Table 6.3 (p 162, Atkins & Carey) shows that this is generally true. (The exceptions, the halogens, are treated in Section 6.10 of Atkins & Carey. We will not discuss them further)
This conclusion also suggests that meta directing groups will also be "deactivating" groups; that is, that benzene rings which carry a meta directing group will be less reactive than those which do not. Table 6.3 also bears this out. Both the meta directing characteristics and the deactivating characteristics are outcomes of the electron withdrawing nature of these groups and the consequent increase in the energy of electrophilic aromatic substitution intermediates when they are present.