
1.8: Enolates, Aldol Condensation, Synthesis
- Page ID
- 81774

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Last time we worked through the reagents which oxidize aldehydes to carboxylic acids and the reagents which reduce aldehydes to primary alcohols and ketones to secondary alcohols. We also learned how enolate ions can be formed by the removal of an alpha hydrogen atom and how these enolate ions can act as nucleophiles toward bromine.
Enols
What happens if we leave out bromine? The alternate electrophile is effectively a H+ from water. It can react either at the alpha carbon (which bears some electron density) or at the oxygen (which also is a basic site). The first reaction just reverses the formation of the enolate, but the second reaction makes a new structure, an enol. Since either reaction produces an OH- ion, either reaction is catalyzed by base.
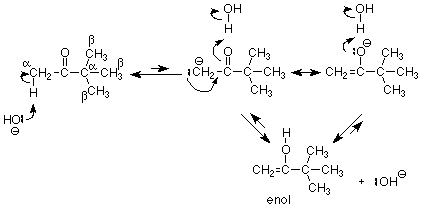
Notice that the product of this mechanism, an enol, is an isomer of the original ketone. It is not a resonance structure, because there is no OH bond in the ketone, so the two structures are differ in how they are connected, and resonance structures may not be that different. Notice also that this is an equilibrium between isomers. Isomers that are in equilibrium with each other are called tautomers. This situation is called keto-enol tautomerism (the same term is used when the carbonyl component is an aldehyde.) Generally, there is very little enol in equilibrium with the keto tautomer, and we will generally write an enol structure when we need it.
Since addition of water was catalyzed by both acid and base, we can ask whether keto-enol tautomerism can also be catalyzed by acid. We know what the first step must be, because an H+ must react with the carbonyl oxygen. This moves electrons away from the carbonyl carbon, which in turn attracts electrons from it's immediate neighborhood, particularly the alpha C-H bond. This bond is thus susceptible to attack by the weakly basic water molecule (there's very little hydroxide ion in an acidic solution) to form the enol.
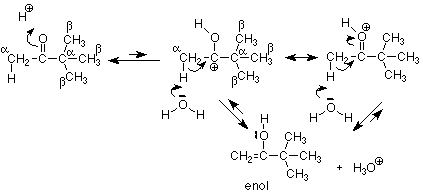
Of course, since a catalyst cannot change the position of equilibrium, the amount of the enol tautomer formed is small whether the reaction is acid catalyzed or base catalyzed.
The term "enol" is a combination of the "ene" ending typical for alkenes (compounds with C=C double bonds) and the "ol" ending typical for alcohols. The anion derived from an enol has the ending "ate" appended as is the case for many polyatomic anions (consider sulfate as an example).
Aldol Addition
One way to describe what we have just done is to say that we have worked out a way to make the alpha-carbon of a carbonyl compound into a carbon nucleophile. Earlier we found that the carbon nucleophile of a Grignard reagent was very important as a synthetic tool. Can we use this alpha-carbon nucleophile similarly? To some extent, the answer is yes. There are a few restrictions:<
- To use the strongest nucleophile we can form at the alpha carbon, we will use base catalysis so as to make enolate ions.
- Since there is little enolate ion at equilibrium, we will use the more reactive carbonyl compound -- an aldehyde, rather than a ketone.
The mechanistic pattern is as follows:
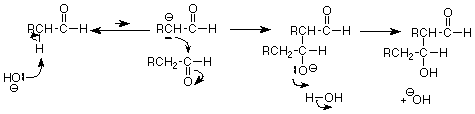
The first step forms the enolate ion. The second step makes a carbon-carbon bond by nucleophilic attack of the enolate alpha-carbon on the carbonyl carbon of a second molecule of the aldehyde. The third step places a proton on the now negatively charged oxygen and regenerates the basic catalyst.
Let's look at the overall reaction and relate the structure of the product to that of the reactant.
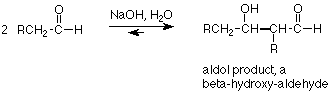
Two molecules of an aldehyde combine so that the alpha-carbon of one becomes attached to the carbonyl carbon of the other. The carbonyl oxygen becomes an alcohol group, located on the beta carbon (relative to the remaining carbonyl group) of the product. The overall reaction is an equilibrium, as we would expect since all steps in our mechanism are equilibria. The equilibrium favors aldol product when the carbonyl group is an aldehyde, but does not for ketones.
If we are seeking to use the aldol addition for synthetic purposes, we look for the characteristic beta-hydroxy-aldehyde arrangement of functional groups. Then we check to see that the R-groups are in fact identical. If these criteria are met, then we can work backwards to the needed aldehyde by mentally breaking the "thick" bond -- the one which was formed in the actual addition step. That gives us our reactant. Incidentally, the term aldol stems from the "ald" of aldehyde and the "ol" of alcohol.
Aldol Condensation
The particular example we have discussed is capable of one more reaction. If we heat the aldol addition product, it usually loses a molecule of water to make a double bond between the alpha and beta carbons of the former aldol. This is a dehydration reaction, and it is also an example of an elimination reaction. An elimination reaction is the structural opposite of an addition reaction, and we'll look at typical mechanisms for these reactions later
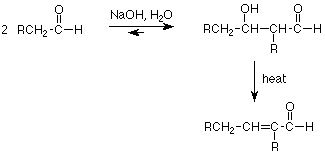
Notice that this elimination reaction requires that the alpha carbon of the aldol product have attached a hydrogen. Since that carbon already lost one hydrogen in forming the enolate intermediate essential to the aldol addition, it must have had two hydrogens in the original aldehyde. The product of this second reaction is called an alpha-beta-unsaturated aldehyde (unsaturated because of the carbon-carbon double bond), and the overall reaction is called an aldol condensation. The term condensation is used to describe a reaction in which two molecules combine to form a larger product with the loss of a molecule of water.
The key structural feature of a molecule which might be made by way of an aldol condensation is the carbon-carbon double bond between the alpha and beta carbons of an aldehyde. If a synthetic target has that feature, we need to check to see if the R-groups are identical. If so, we can make that molecule by the aldol condensation.
In your later biochemical studies you will find that carbon-carbon bonds are often made (in photosynthesis for example) by enzymatically catalyzed aldol additions. The reverse reaction is also common (called a retro-aldol reaction), particularly in glycolysis, the metabolism of glucose.