
11.7: Oxidation Reactions
- Page ID
- 22257

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Ozonization
Most alkenes react readily with ozone \(\left( \ce{O_3} \right)\), even at low temperatures, to yield cyclic peroxidic derivatives known as ozonides. For example,
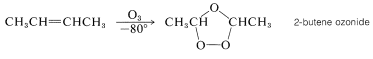
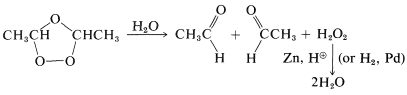
These substances, like most compounds with peroxide \(\left( \ce{O-O} \right)\) bonds, may explode violently and unpredictably. Therefore ozonizations must be carried out with appropriate caution. The general importance of these reactions derives not from the ozonides, which usually are not isolated, but from their subsequent products. The ozonides can be converted by hydrolysis with water and reduction, with hydrogen (palladium catalyst) or with zinc and acid, to carbonyl compounds that can be isolated and identified. For example, 2-butene gives ethanal on ozonization, provided the ozonide is destroyed with water and a reducing agent which is effective for hydrogen peroxide:
An alternative procedure for decomposing ozonides from di- or trisubstituted alkenes is to treat them with methanol \(\left( \ce{CH_3OH} \right)\). The use of this reagent results in the formation of an aldehyde or ketone and a carboxylic acid:
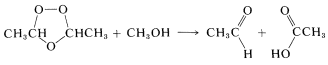
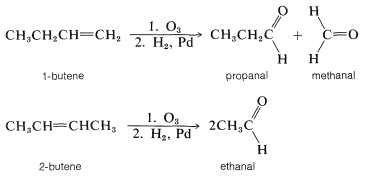
The overall ozonization reaction sequence provides an excellent means for locating the positions of double bonds in alkenes. The potentialities of the method may be illustrated by the difference in reaction products from the 1- and 2-butenes:
Mechanism of Ozonization
Ozonization of alkenes has been studied extensively for many years, but there is still disagreement about the mechanism (or mechanisms) involved because some alkenes react with ozone to give oxidation products other than ozonides. It is clear that the ozonide is not formed directly, but by way of an unstable intermediate called a molozonide. the molozonide then either isomerizes to the "normal" ozonide or participates in other oxidation reactions. Although the structure of normal ozonides has been established beyond question, that of the molozonide, which is very unstable even at \(-100^\text{o}\), is much less certain.
The simplest and most widely accepted mechanism involves formation of a molozonide by a direct cycloaddition of ozone to the double bond.\(^1\)
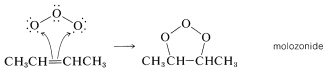
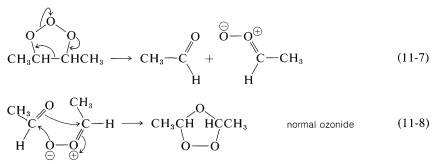
Isomerization of the molozonide appears to occur by a fragmentation-recombination reaction, as shown in Equations 11-7 and 11-8:
Hydroxylation of Alkenes
Several oxidizing reagents react with alkenes under mild conditions to give, as the overall result, addition of hydrogen peroxide as \(\ce{HO-OH}\). Of particular importance are alkaline permanganate \(\left( \ce{MnO_4^-} \right)\) and osmium tetroxide \(\left( \ce{OsO_4} \right)\), both of which react in an initial step by a suprafacial cycloaddition mechanism like that postulated for ozone.
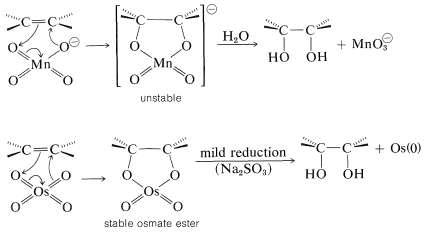
Each of these reagents produces cis-1,2-dihydroxy compounds (diols) with cycloalkenes:
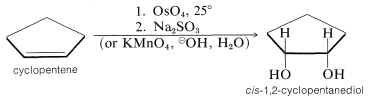
Osmium tetroxide is superior to permanganate in giving good yields of diol, but its use is restricted because it is a very costly and very toxic reagent.
Oxidation with Peroxidic Compounds. Oxacyclopropane (Oxirane) Formation
Alkenes can be oxidized with peroxycarboxylic acids, \(\ce{RCO_3H}\), to give oxacyclopropanes (oxiranes, epoxides), which are three-membered cyclic ethers:
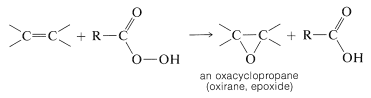
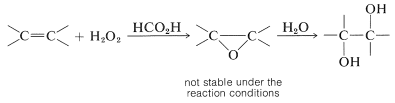
The reaction, known as epoxidation, is valuable because the oxacyclopropane ring is cleaved easily, thereby providing a route to the introduction of many kinds of functional groups. In fact, oxidation of alkenes with peroxymethanoic acid (peroxyformic acid), prepared by mixing methanoic acid and hydrogen peroxide, usually does not stop at the oxacyclopropane stage, but leads to ring-opening and the subsequent formation of a diol:
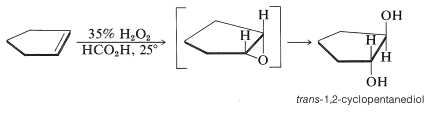
This is an alternative scheme for the hydroxylation of alkenes (see Section 11-7C). However, the overall stereochemistry is opposite to that in permanganate hydroxylation. For instance, cyclopentene gives trans-1,2-cylcopentanediol. First the oxirane forms by suprafacial addition and then undergoes ring opening to give the trans product:
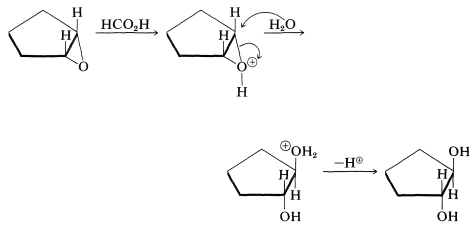
The ring opening is a type of \(S_\text{N}2\) reaction. Methanoic acid is sufficiently acidic to protonate the ring oxygen, which makes it a better leaving group, thus facilitating nucleophilic attack by water. The nucleophile always attacks from the side remote from the leaving group:
The peroxyacids that are used in the formation of oxacyclopropanes include peroxyethanoic \(\left( \ce{CH_3CO_3H} \right)\), peroxybenzoic \( \left( \ce{C_6H_5CO_3H} \right)\), and trifluoroperoxyethanoic \(\left( \ce{CF_3CO_3H} \right)\) acids. A particularly useful peroxyacid is 3-chloroperoxybenzoic acid, because it is relatively stable and is handled easily as the crystalline solid. The most reactive reagent is trifluoroperoxyethanoic acid, which suggests that the peroxyacid behaves as an electrophile (the electronegativity of fluorine makes the \(\ce{CF_3}\) group strongly electron-attracting). The overall reaction can be viewed as a cycloaddition, in which the proton on oxygen is transferred to the neighboring carbonyl oxygen more or less simultaneously with formation of the three-membered ring:
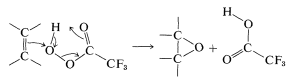
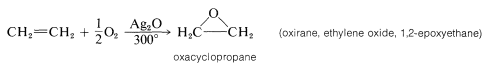
A reaction of immense industrial importance is the formation of oxacyclopropane itself (most often called ethylene oxide) by oxidation fo ethene with oxygen over a silver oxide catalyst at \(300^\text{o}\):
Oxacyclopropane is used for many purposes, but probably the most important reaction is ring opening with water to give 1,2-ethanediol (ethylene glycol, bp \(197^\text{o}\)). This diol, mixed with water, is employed widely in automotive cooling systems to provide both a higher boiling and lower freezing coolant than water alone:
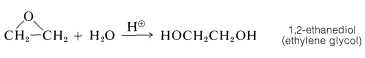
Propene and higher alkenes are not efficiently epoxidized by oxygen and \(\ce{Ag_2O}\) in the same way as ethene because of competing attack at other than the double-bond carbons.
\(^1\)The ozone structure shown here with single electrons having paired spins on the terminal oxygens accords both with the best available quantum mechanical calculations and the low dipole moment of ozone, which is not consonant with the conventional \(\ce{O=} \overset{\oplus}{\ce{O}} - \overset{\ominus}{\ce{O}}\) structure. See W. A. Goddard III, T. H. Dunning, Jr., W. J. Hunt, and P. J. Hay, Accounts of Chemical Research 6, 368 (1973).
Contributors and Attributions
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."