
11.3: Properties of Alcohols and Phenols
- Page ID
- 227131
After completing this section, you should be able to
- explain why the boiling points of alcohols and phenols are much higher than those of alkanes, ethers, etc., of similar molecular mass.
- discuss the factors that are believed to determine the acidity of alcohols and phenols.
- list a given series of alcohols or phenols in order of increasing or decreasing acidity.
- explain the difference in acidity between two given alcohols or phenols.
- explain why phenols are more acidic than alcohols.
- explain, in terms of inductive and resonance effects, why a given substituted phenol is more or less acidic than phenol itself.
- write equations for the reactions of given alcohols and phenols with strong bases, such as sodium hydride and sodium amide.
Make certain that you can define, and use in context, the key terms below.
- acid ionization constant (Ka)
- alkoxide ion (RO−)
- phenoxide ion (ArO−)
You may wish to review the concept of hydrogen bonding, which should have been discussed in your first‑year general chemistry course.
Boiling Points of Alcohols
The oxygen in alcohols and phenols is sp3 hybridized which gives the roughly the same tetrahedral geometry as water. The

The presence of a highly electronegative oxygen confers a measure of polar character to alcohols. Much of the electron density of an alcohol is drawn towards the oxygen, giving alcohols a relatively high dipole momeny (1.7 D for Methanol).
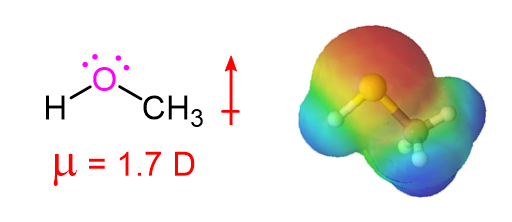
The Dipole Moment of Methanol
Alcohols contain an extremely polar covalent O-H bond. The electrons in this bond are strongly drawn towards the oxygen giving it a partial negative charge (delta-) while the electrons are drawn away from the hydrogen giving is a partial positive charge (delta+). The polar character of this bond leads to an interaction between alcohol molecules where the positively polarized hydrogen of one alcohol molecule is attracted to the lone pair electrons on the electronegative oxygen atom of a different alcohol molecule. This creates a type of intramolecular attraction is called “hydrogen bonding.” Although the strength of such attractions are much less than most conventional chemical bonds, they are still significant (about 5 to 10 kcal per bond). For an alcohol to vaporize, these intramolecular hydrogen bonding attractions must be broken making alcohols substantially less volatile, have higher boiling points, and greater water solubility than molecules with a similar molecular weight (See Table 1 below). An example of this is seen when comparing 1-propanol (MW = 60.1), Chloroethane (MW = 64.5), and butane (MW = 58.1) which have boiling points of 94.7 oC, 12.3 oC, and -1 oC respectively.
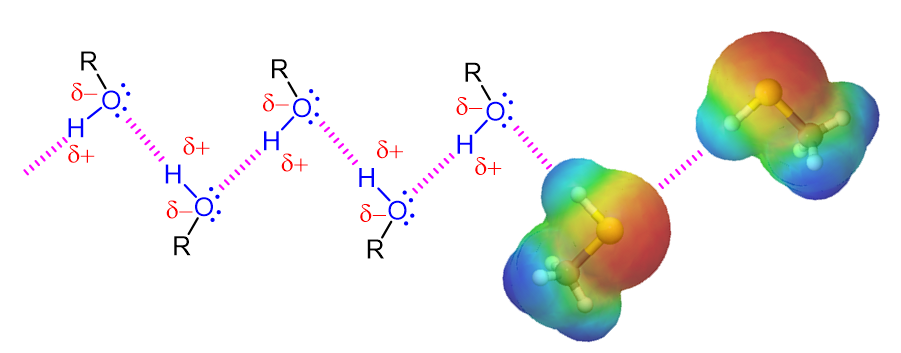
A Representation of Hydrogen Bonding in Alcohols
This table shows that alcohols (in blue) have higher boiling points than haloalkanes and alkanes with the equivalent molecular weight. It also shows that the boiling points of alcohols increase with the number of carbon atoms.
| Compound | IUPAC Name | Molecular Weight (g/mol) | Melting Point (oC) | Boiling Point (oC) |
|---|---|---|---|---|
| CH3OH | Methanol | 32.0 | -97.8 | 65.0 |
| CH3Cl | Chloromethane | 50.5 | -97.7 | -24.2 |
| CH3CH2OH | Ethanol | 46.1 | -114.7 | 78.5 |
| CH3CH2CH2CH3 | Butane | 58.1 | -140. | -1 |
| CH3CH2CH2OH | 1-Propanol | 60.1 | -126.5 | 97.4 |
| CH3CH2Cl | Chloroethane | 64.5 | -136.4 | 12.3 |
| CH3CH2CH2CH2CH3 | Pentane | 72.2 | -130 | 36.3 |
| CH3CH2CH2CH2OH | 1-Butanol | 74.1 | -89.5 | 117.3 |
| CH3(CH2)4OH | 1-Pentanol | 88.1 | -79 | 138 |
Solubility of Alcohols in Water
Alcohols and water have the ability to form hydrogen bonds with one another which tends to make the two liquids miscible. Small alcohols are completely soluble in water; mixing the two in any proportion generates a single solution. However, solubility decreases as the length of the hydrocarbon chain in the alcohol increases. At four carbon atoms and beyond, the decrease in solubility is noticeable; a two-layered substance may appear in a test tube when the two are mixed.
Basic Properties of Alcohols
Alcohols are weak bases similar in strength to water and can accept protons from strong acids to form the conjugate acid called oxonium ions (ROH2+). An example is the reaction of methanol with hydrogen bromide to give methyloxonium bromide, which is analogous to the formation of hydroxonium bromide from the reaction of hydrogen bromide and water:
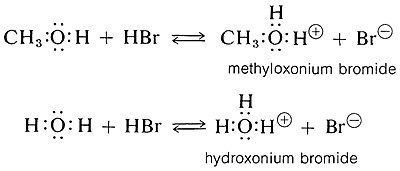
Acidic Properties of Alcohols
Alcohols, like water, are both weak bases and weak acids. In aqueous solutions, alcohols dissociate slightly by donating a hydrogen to water. This creates the alcohol's conjugate base, called an alkoxide ion (RO-), along with hydronium (H3O+). The acid ionization constant (Ka) of ethanol is about 1018, which is slightly less than that of water. Alcohols, such as ethanol, can be deprotonated to form its conjugate base by reaction with a stronger base, such as sodium amide (NaNH2), sodium hydride (NaH), or Grignard reagents (RMgBr). Alkoxides can also be formed using sodium or potassium metal which reacts vigorously but controllably with alcohols.

Once created, alkoxides act as bases and are often used as reagents in organic chemistry. Alkoxide's systematic names are based of the corresponding alcohol with the suffix -ate added. For example, ethanol becomes ethanolate. They are named systematically by adding the -ate suffix to the name of the alcohol. Methanol becomes methanolate, for instance. Common names are often used for small alkoxides. Common names are generated as: (Name of the positive counter ion) (Prefix for the parent chain + oxide). Example: ethanol becomes ethoxide.

As discussed in Section 2.8, the strength of an acid in water can be expressed either by its Ka or its pKa. Compounds with a lower Ka and a higher pKa are considered less acidic than compound with a higher Ka and a lower pKa.
\[ K_a = \dfrac{[A^-][H_3O^+]}{[HA]} \label{First} \]
\[pK_a = -\log K_a \nonumber \]
In general, alcohols in aqueous solution are slightly less acidic than water. The order of acidity of various liquid alcohols generally is water > primary > secondary > tertiary ROH. By this we mean that the pKa is reduced as R is changed from primary to secondary to tertiary; therefore, tert-butyl alcohol is less acidic than ethanol. This trend is explained by the importance of solvation in equilibrium. In solution, the larger alkoxide ions, are less well solvated than the smaller ions, because fewer solvent molecules can be accommodated around the negatively charged oxygen in the larger ions. Acidity of alcohols therefore decreases as the size of the conjugate base increases. This trend can be clearly seen when comparing alkoxide size to the pKa of the corresponding alcohol listed in Table 2.

\[ ROH + OH^- \rightleftharpoons RO^- + HOH \nonumber \]
| R | Name | pKa1 |
|---|---|---|
| H | water | 14.0 |
| CH3 | methanol | 15.5 |
| CH3CH2 | ethanol | 15.9 |
| (CH3)2CH | propan-2-ol (isopropyl alcohol) | 16.5 |
| (CH3)3C | 2-methylpropan-2-ol (tert-butanol) | 17 |
| C6H5 (phenyl) | phenol | 9.95 |
The addition of an electron-withdrawing group, such as an electronegative halogen, can increase the acid strength of an alcohol by stabilizing its alkoxide conjugate base through induction (Section 2.10). The electron-withdrawing group helps to spread out the electron density of the alkoxide's negative charge, which has a stabilizing effect. The inductive effect is cumulative such that the acid strength of an alcohol becomes stronger (Lower pKa) as the number of halogens increases. The presence of nine fluorines in nonafluoro-tert-butyl alcohol decreases its pKa to 5.4 which is significantly more acidic than tert-butyl alcohol (pKa = 18). The electron-withdrawing effect of the fluorines is clearly seen when comparing the electrostatic potential maps of the corresponding alkoxides. In tert-butoxide the molecule's electron density is firmly centered around the oxygen as shown by the orange/yellow color. In nonafluoro-tert-butoxide the molecule's electron density is almost completely removed from the oxygen and shifted to the fluorines.
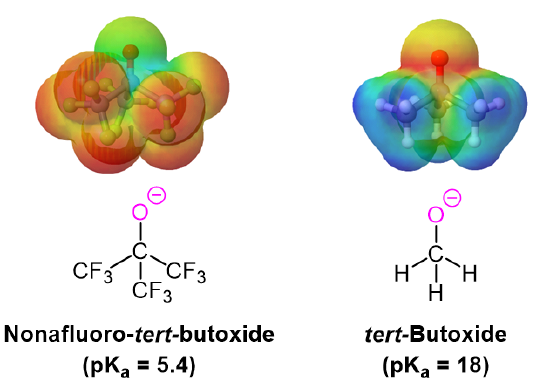
Acidity of Phenol
Another method for increasing acidity is through stabilizing a conjugate base through resonance effects. An excellent example of this effect is shown through phenol being roughly a million times more acidic than cyclohexanol.
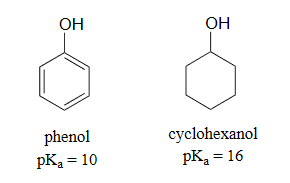
Phenol is acidic enough to be deprotonated by weaker bases, such as sodium hydroxide (NaOH), to form the phenoxide ion.

The increased acidity of phenol is caused by the negative charge and a set of lone pair electrons from the phenoxide's oxygen atom being delocalized by resonance to three different carbons on the aromatic ring. As a result, the negative charge is no longer entirely localized on the oxygen, but is spread throughout the whole ion allowing it to be highly stabilized.
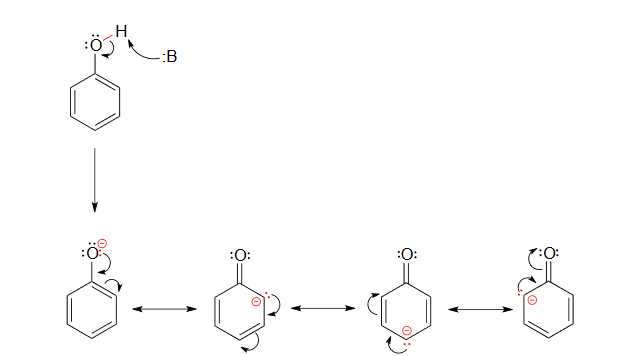
Although these are all minor resonance contributors (negative charge is placed on a carbon rather than the more electronegative oxygen), they nonetheless have a significant effect on the acidity of the phenolic proton. Essentially, the benzene ring is acting as an electron-withdrawing group by resonance.
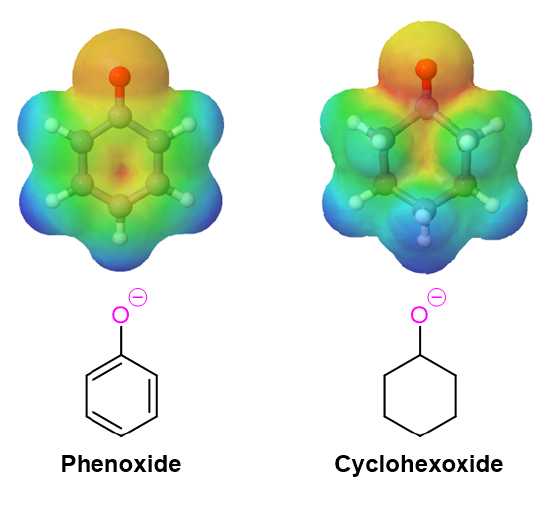
The electrostatic potential maps below compare the phenoxide ion to an unconjugated alkoxide. The negative charge of the phenoxide ion, show as a yellow/orange color, is delocalized from oxygen into the aromatic ring (seen by the growing red color in the phenoxide ring).
Acidity of Substituted Phenols
Phenolic acidity is further enhanced by the presence of an additional electron-withdrawing substituent, such as a nitro or carbonyl, on the aromatic ring. For the conjugate base of the phenol derivative below, an additional resonance contributor can be drawn in which the negative formal charge is placed on the carbonyl oxygen.

Now the negative charge on the conjugate base can be spread out over two oxygens (in addition to three aromatic carbons). The phenol acid therefore has a pKa similar to that of a carboxylic acid, where the negative charge on the conjugate base is also delocalized to two oxygen atoms. The ketone group on the aromatic ring is acting as an electron withdrawing group and 'pulling' electron density towards itself, through both inductive and resonance effects.
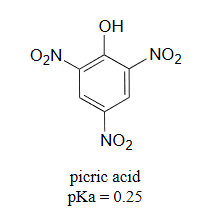
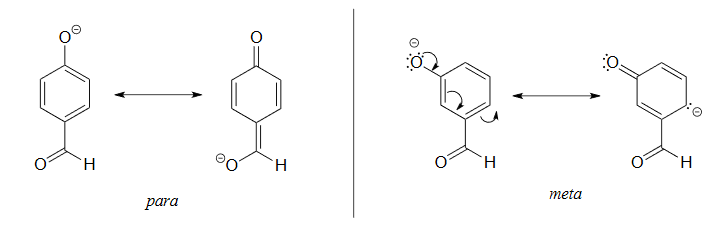
It is noteworthy that the influence of a nitro substituent is over ten times stronger in the para-location than it is meta, despite the fact that the latter position is closer to the hydroxyl group. This occurs since nitro groups at the meta position cannot accept the negative charge through resonance. Furthermore additional nitro groups have an additive influence if they are positioned ortho or para locations to the hydroxide. The trinitro compound shown at the lower right is a very strong acid called picric acid.
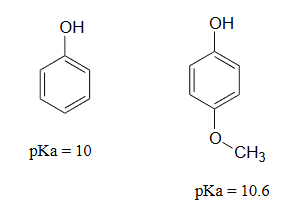
Lastly, if an electron donating group is attached to aromatic ring, as in p-methoxyphenol, the phenoxide ion is destabilized which causes a decrease in acidity in the corresponding phenol.
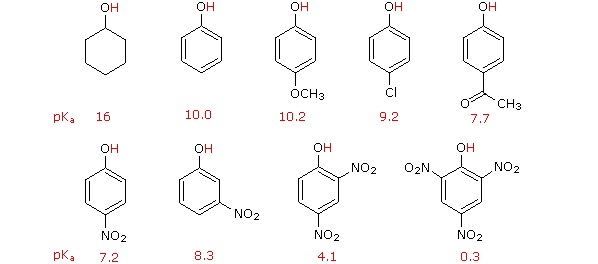
Using resonance structures, please explain why 4-methoxyphenol is less acidic than phenol.
- Answer
-
The methoxy group is an electron-donating group by resonance. A resonance contributor can be drawn in which a formal negative charge is placed on the carbon adjacent to the negatively-charged phenolate oxygen.
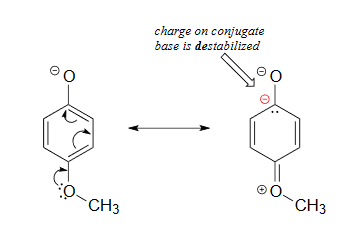
Because of like-charge repulsion, this destabilizes the negative charge on the phenolate oxygen, making the corresponding phenol less acidic.
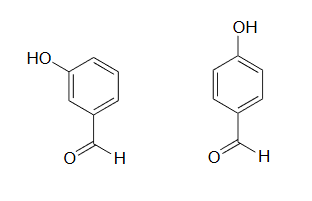
1) The position of the electron-withdrawing substituent relative to the phenol hydroxyl is very important in terms of its effect on acidity. Which of the two substituted phenols below is more acidic? Use resonance drawings to explain your answer.
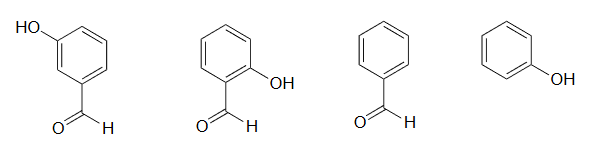
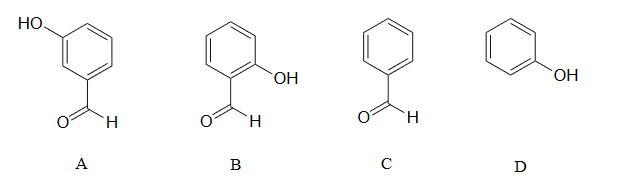
2) Rank the four compounds below from most acidic to least.
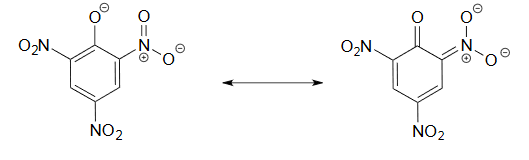
3) Use a resonance argument to explain why picric acid has such a low pKa.
4) Predict which compound of each pair is more soluble in water and explain your reasoning.
a) Butan-1-ol or pentan-1-ol
b) Phenol or cyclohexanol
c) Octan-1,3-diol or octan-1-ol
d) 1-Chlorohexane or hexan-1-ol
5) Predict which compound has the higher boiling point and explain your reasoning.
a) Water or ethanol
b) Butan-1-ol or octan-1-ol
c) Hexan-2-ol or hexan-2-one
- Answer
-
1) The para-subsituted phenol is more acidic, because the negative charge on the conjugate base can be delocalized to the aldehyde oxygen. This is not possible when the aldehyde group is in the meta position
2) Compound B is the strongest acid due to electron-withdrawing resonance effects-the negative charge on the conjugate base can be delocalized to the aldehyde oxygen. Compound A is the 2nd strongest acid -while the negative charge on the conjugate base cannot be delocalized to the aldehyde oxygen due to the meta-position, the aldehyde none the less has a stabilizing, electron-withdrawing inductive effect. Compound D is ranked #3 -it is phenol, and does not have any electron-withdrawing substituents on the ring as do B and A. Compound C is the least acidic -neither the phenyl group nor the aldehyde are even slightly acidic.
3) The negative charge on the conjugate base of picric acid can be delocalized to oxygen atoms on all threeof the nitro groups. One such resonance contributor is shown below. This extensive delocalization means that the conjugate base is very stable, and the conjugate acid is thus a very strong acid.
4) a) Butan-1-ol is more soluble in water because it has a smaller hydrophobic region compared to pentan-1-ol, allowing butan-1-ol to interact with water better.
b) Phenol is more soluble in water than cyclohexanol because of the more polar character of its ring. phenol is able to interact with water better than cyclohexanol due to the conjugated pi-system of electrons in its ring, which which gives it a more ionic character.
c) Octan-1,3-diol is more soluble in water as it has two hydroxy groups, allowing it to form more hydrogen bonds and interact with water better than octan-1-ol.
d) Hexan-1-ol is more soluble in water as it can hydrogen bond compared to alkyl halides, such as 1-chlorohexane, which are insoluble in water.
5) a) Water has a higher boiling point compared to ethanol as it participates in more hydrogen bonding with other water molecules, thus requiring more energy to break the intermolecular attractions between water molecules.
b) Octan-1-ol has the higher boiling point compared to butan-1-ol. Both alcohols can H-bond, however the longer hydrophobic carbon chain tail of octan-1-ol experiences more van der Waal interactions compared to the shorter hydrophobic region of butan-1-ol leading to a higher boiling point.
c) Since hexan-1-ol can H-bond, it has a higher boiling point than hexan-2-one, which cannot H-bond.