
5.5: Oxidative Addition
- Page ID
- 83500
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)How important are oxidative additions of non-polar reagents? Very. The addition of dihydrogen (H2) is an important step in catalytic hydrogenation reactions. Organometallic C–H activations depend on oxidative additions of C–H bonds. In a fundamental sense, oxidative additions of non-polar organic compounds are commonly used to establish critical metal-carbon bonds. Non-polar oxidative additions get the ball rolling in all kinds of catalytic organometallic reactions. Here the mechanisms and important trends associated with non-polar oxidative additions are discussed.
Oxidative Additions of H2
Electron-rich metal centers with open coordination sites (or the ability to form them) undergo oxidative additions with dihydrogen gas. The actual addition step is concerted, as we might expect from the dull H2 molecule! However, before the addition step, some interesting gymnastics are going on. The status of the σ complex that forms prior to H–H insertion is an open question—for some reactions it is a transition state, others a discrete intermediate. In either case, the two new hydride ligands end up cis to one another. Subsequent isomerization may occur to give a trans dihydride.

Oxidative addition of dihydrogen to Vaska’s complex. Note the cis arrangement of the hydride ligands.
There’s more to this little reaction than meets the eye. For starters, either pair of trans ligands in the starting complex (L/L or Cl/CO) may “fold back” to form the final octahedral complex. As in associative ligand substitution, the transition state for folding back is basically trigonal bipyramidal. As we saw before, π-acidic ligands love the equatorial sites of the TBP geometry, which are rich in electrons capable of π bonding. As a consequence, π-acidic ligands get folded back preferentially, and tend to end up cis to their trans partners in the starting complex.

Dihydrogen may approach along two distinct trajectories. Placing π-acidic ligands in the equatorial plane of the TBP transition state is favored.
Termolecular oxidative additions of H2, in which the two H atoms find their way to two different metal centers, are also known, but these suffer from entropic issues, since H2 is jammed between two metal complexes in the transition state of the (concerted) mechanism.
Oxidative Additions of Silanes (H–Si)
Silanes bearing Si–H bonds may react with organometallic complexes in oxidative addition reactions. Spectroscopic experiments support the intermediacy of a silyl σ complex before insertion. Since the mechanism is concerted, oxidative addition occurs with retention of configuration at Si. The usual pair of forward bonding (σSi–H→dσ) and backbonding (dπ→σ*Si–H) orbital interactions are at play here. File this reaction away as a great method for the synthesis of silyl complexes.
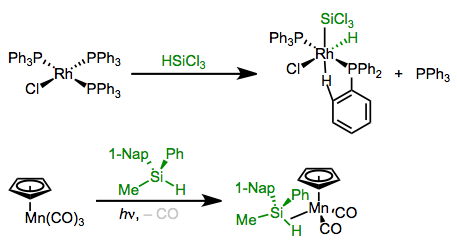
Si–H bonds undergo oxidative addition to electron-rich metal complexes. Electron-poor complexes may stop at the σ complex stage.
Oxidative Additions of C–H Bonds
Needless to say, oxidative addition reactions of C–H bonds are highly prized among organometallic chemists. As simple as it is to make silyl complexes through oxidative addition, analogous reactions of C–H bonds that yield alkyl hydride complexes are harder to come by.
The thermodynamics of C–H oxidative addition tell us whether it’s favorable, and depend heavily on the nature of the organometallic complex. The sum of the bond energies of the new M–C and M–H bonds must exceed the sum of the energies of the C–H bond and any M–L bonds broken during the reaction (plus, OA is entropically disfavored). For many complexes, the balance is not in favor of oxidative addition. For example, the square planar Vaska’s complex (L2(CO)IrCl; L = PPh3) seems like a great candidate for oxidative addition of methane—at least to the extent that the product will be six-coordinate and octahedral. However, thermodynamics is a problem:
104 (C–H) – [60 (Ir–H) + 35 (Ir–Me)] + 9 kcal/mol (entropy) = 18 kcal/mol
18 kcal/mol is prohibitively high in energy, and playing with the temperature to adjust the entropy factor can’t “save” the reaction.
More electron-rich complexes exhibit favorable thermodynamics for insertions of C–H bonds. The example below is so favorable (104 – [75 + 55] + 9 = –17 kcal/mol) that the product is a rock!
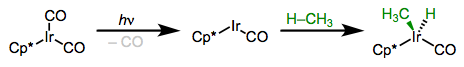
This thermodynamically favorable C–H oxidative addition is helped by the electron-donating Cp* ligand.
Arenes undergo C–H oxidative addition faster (and more favorably) than alkanes for several reasons. It seems likely that an intermediate arene π complex and/or C–H σ complex precede insertion, and these complexes ought to be more stable than alkyl σ complexes. In addition, metal-aryl bonds tend to be stronger than metal-alkyl bonds.
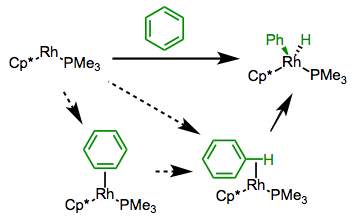
Mechanistic possibilities for the oxidative addition of arene C–H bonds.
Your average organic compound is covered in C–H bonds. Thus, for C–H oxidative addition to be synthetically useful, we need to understand how to control the selectivity of the reaction. In most early studies, selectivity for insertion into primary C–H bonds was observed. It became apparent that primary alkyl complexes (viz., the observed products) are generally much more stable than secondary alkyl complexes, even though the corresponding primary and secondary σ complexes are comparable in energy. Steric factors are a likely culprit!
Naturally, other strategies for controlling selectivity in C–H “activations” have appeared; however, be aware that these may not involve oxidative addition. Lewis basic directing groups have been used with success to direct oxidative addition to a nearby C–H bond—but these reactions are aided by base and do not involve oxidation. Metal carbenoid insertions into C–H bonds also do not involve oxidation at the metal center. The lesson of these reactions: be careful, and don’t assume that all alkyl complexes are the result of C–H oxidative addition!