
Theory
- Page ID
- 60819

Let us examine the nature of the double-layer capacitance first before discussing the electron transfer reaction. The current (i.e., electrons) flows to the working electrode (WE) in order to bring its potential to some desired value. A potentiostat with a 3-electrode cell provides the current via the auxiliary electrode (AE) to the WE while the potential is measured with respect to a reference electrode (RE). Thus, the current can be ascribed wholly to that which flows to change the potential of the WE from Ei to Ef with respect to RE. It is instructive to model this situation as being analogous to charging a capacitor, as illustrated in Figure 1. In this circuit, a resistor, R, represents the cell resistance. It is placed in series with a capacitor (C) that represents the electrode double-layer capacitance. A switch, SW, when closed, imposes the voltage of a battery, Vb, to the circuit. Two voltage-measuring devices monitor the voltages, VR and VC, across the resistor and the capacitor, respectively.
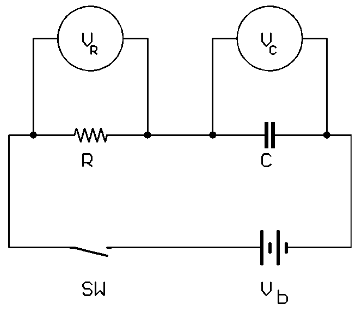
Figure 1.
At open circuit (i.e., time t = 0), prior to the closure of SW, VR and VC = 0.0 volt. When SW is closed, the instantaneous value of VR ⇒ Vb and the instantaneous (i.e., initial current) current, io, is given by: io = Vb/R. That is, the entire Vb is imposed across R since there is no charge yet residing on the capacitor. The current, after its initial rise, decays exponentially as a function of time: it = io exp (-t/RC). This it–t profile is shown in Figure 2. The voltage of the capacitor rises exponentially with time - VC = Vb [1 – exp (-t/RC)]. At t => ∞, VR = 0 and VC = Vb. Thus, VC vs. t is a mirror image of it vs. t.
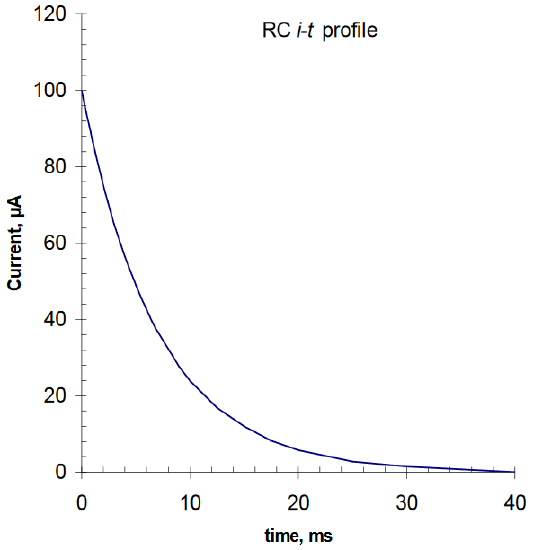
Figure 2. Calculated chronoamperometry i vs. t profile for Vb = 1.0 volt, R = 10,000 Ω and C = 2 μF with a 3 mm diameter disk electrode.
In a real electrochemical cell, the value of R is usually much smaller and the value of C depends on the type and area of the electrode (Pt, Au, GC or other material) and the solution condition. That is, the type of solvent, and the type and concentration of electrolyte. Nominal values of C are in the range of 10 – 20 μF/cm2 for most electrodes.
Often, the i-E wave obtained with cyclic voltammetry is used to calculate C. That is, the charge, q, is given by: q = it = CAE where E is the potential of the electrode (vs. reference electrode), A is the area, and the other terms have their usual meaning. Differentiating the equation with respect to time, t, and rearranging terms, one obtains: i = CA(dE/dt) where dE/dt is the scan rate. The relationship assumes that the capacitance remains constant independent of the applied potential; i.e., (dC/dE) = 0. Such is seldom the case since nearly all electrodes undergo some finite surface reaction that consumes charge. However, a good estimate of C can be obtained by scanning the electrode over a relatively narrow potential window. The C value can be calculated from the i-E envelope.
Let us examine now a current, I, vs. time t, response in the presence of an electroactive species that undergoes an electron transfer reaction at a diffusion-controlled rate. Under these conditions, the current is given by the Cottrell relationship:
\[i = \dfrac{n\,\mathrm{F\,A\,D}^{1/2}C^b}{(\pi\, t)^{1/2}}\tag{1}\]
where n is the number of electron(s) transferred per electroactive molecule or ion; F is Faraday's constant; A is the area of the electrode surface in cm2; D is the diffusion coefficient in cm2/s; Cb is the concentration of the electroactive species in mol/cm3; and t is time in second. The current rises rapidly to a maximum value when the potential goes from its initial value, Ei, where reaction occurs, to its final value, Ef, and then decays as a function of t1/2, as seen in Figure 3.
The value of it1/2 as a function of t should be constant. The value of nD1/2 can be determined from this value, assuming that A (the area) and Cb (bulk concentration) are known.
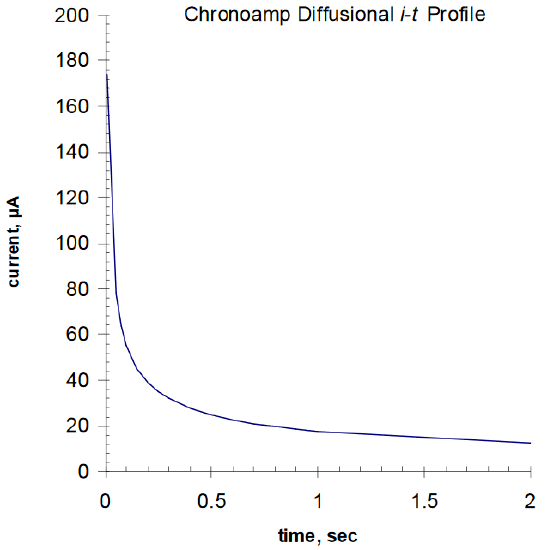
Figure 3. Cottrell Diffusion i-t profile for 3 mM disk electrode in 1 mM ferricyanide.
What should be the values of the initial, Ei, and final potential, Ef? Ei should be at a value where no electrode reaction occurs. You can decide what this value should be by looking at a cyclic voltammogram (see Experiment titled: Cyclic Voltammetry at Solid Electrodes for directions). Ef is set at a value that is 0.120 to 0.180 V greater than the reversible potential, Eo. For example, in the case of ferri/ferrocyanide:
\[\mathrm{Fe(CN)_6^{-3}+ e^- == Fe(CN)_6^{-4} \hspace{30px} E^o = 0.36\: V\: vs.\: NHE}\tag{2}\]
Values of Ef can be calculated
\[\mathrm{E_f = E^o -0.120\: V = 0.24\: V\: for\: reduction\: of\: Fe(CN)_6^{-3}\: to\: ferrocyanide.} \tag{3}\]
or
\[\mathrm{E_f= E^o + 0.120\: V = 0.48\: V\: for\: oxidation\: of\: Fe(CN)_6^{-4}\: to\: ferricyanide.} \tag{4}\]
Please remember to correct these potentials for the type of reference electrode you are using!
Think about why the potential is moved to more positive values when you want to remove electrons and to more negative values when you want to add electrodes to a species.
The current is a composite of the double-layer charging and the faradaic current - the latter due to the electron transfer reaction with an electroactive species. The former current decays much more rapidly as an exponential function compared to the square root of time for a diffusion-controlled reaction.
It is instructive to have a mental picture of the diffusional profile of the oxidized and reduced forms of the electroactive species for a CA experiment. If the concentration of the reduced species is 1 mM {CR (t = 0) = 1 mM} before the potential is applied and the concentration of the oxidized species is zero {COX (t=0) = 0}, these concentrations are reversed in the solution phase adjacent to the electrode when Ef is applied. That is, R is oxidized to OX, the ratio of COX/CR => 100/1, assuming Ef changes by +120 mV from Ei at t > 0. The depletion of CR at the electrode causes R species to be transported from the bulk solution to the electrode and OX to be transported out as the electrolysis proceeds. A concentration gradient of these respective species is created, extending further into the bulk solution with time. Graphic illustrations of gradient profiles are illustrated in the website:
www.epsilon-web.net/Ec/manual...hronol/ca.html.
Scroll down to “Analysis of Current Response” to see these profiles.
How far into the solution does this concentration gradient, δ, or diffusion layer extend? It depends on the time and the value of the diffusion coefficient, D, or the designated DR and DOX, the reduced and oxidized species, respectively. The computed values of δ vs. t are for ferri-/ferrocyanide (D value of 0.65 x 10-5 cm2/s at T = 25oC) as plotted in Figure 4. The thickness changes very rapidly initially and then decreases more slowly with time, but overall, δ is small1 (e.g., 0.0052 cm at 10 second).
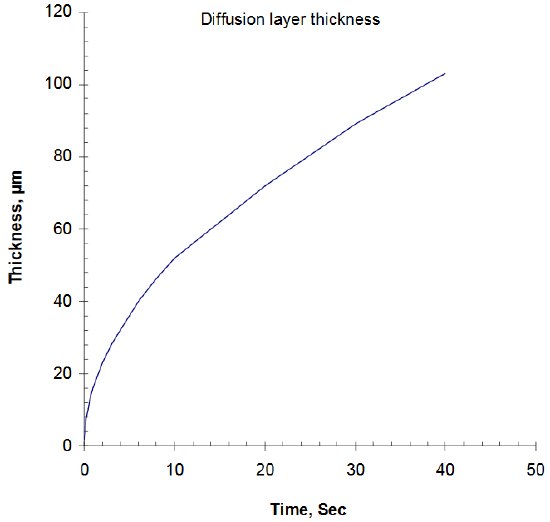
Figure 4. Diffusion layer thickness as a function of time for ferricyanide reduction.
Sample Calculation: Figure 3 gave an illustrative Cottrell current as a function of time. Calculate and plot values of this current for the following experimental conditions and include them in your laboratory report.
n = 1
A = 0.0706 cm2 (for a 3 mM diameter planar electrode)
D = 0.65 x 10-5 cm2/s
CR = 1.0 mM (t = 0, x = 0 to ∝)
COX = 0.0 mM (t = 0, x = 0 to ∝)
F = 96,486 coulomb/mole
1 An approximate value of δ is given by: δ = (2/π) (Dt)1/2 = 0.69 (Dt)1/2
A famous chemist once said: things move approximately 1 cm/day in aqueous solution. Why only 1 cm/day? Remember that the solution is quiet, not stirred, without any convection. Most D values lie in the range of 1.0 x 10-5 to 0.2 x 10-5 cm2/sec (the cm2 comes from the area of a planar electrode).