
IB2. Proteins
- Page ID
- 4056

IB2. Intermolecular Forces in Biology: Proteins (contributed by Henry Jakubowski)
Proteins are a large biological molecules that have molecular weights ranging from the thousand to the millions. Humans have about 24,000 different proteins which catalyze chemical reactions, recognize foreign molecules and pathogens, allow cellular and organism movement, and regulate cell response, including cell division and death.
Proteins are polymers consisting of monomers call amino acids. There are twenty different naturally occurring amino acids that differ in one of the four groups connected to a central carbon atom. In an amino acid, the central (alpha) carbon has an amine group (RNH2, RNH3+), a carboxylic acid group (RCOOH, RCOO-), an H, and one of twenty different R groups (also called side chains) attached to it. The R groups are classified as generally nonpolar, polar charged, or polar uncharged. The smallest amino acid is glycine (Gly) which has a hydrogen atom as its R group. All of the other 19 naturally-occurring amino acids have one stereocenter at the carbon containing the amine and carboxyl groups and can exist as two possible enantiomers of which only one form occurs in proteins. All amino acids in proteins have the absolute configuration shown below. With the exception of the amino acid cysteine (Cys) with a -CH2SH for an R group (which happens to have an R stereocenter), all of the remaining amino acids found in proteins have an S stereocenter.

Amino acids form polymers when an amino group of an amino acid is covalently attached to the carbonyl carbon (C=O) of the carboxyl group of the next amino acid. The resulting link between the amino acids is an amide bond which biochemists call a peptide bond. In this reaction, water is released. In a reverse reaction, the peptide bond can be cleaved by water (hydrolysis). When two amino acids link together to form an amide link, the resulting structure is called a dipeptide. Likewise, we can have tripeptides, tetrapeptides, and other polypeptides. At some point, when the structure is long enough, it is called a protein. There are many different ways to represent the structure of a polypeptide or protein. Each shows differing amounts of information. A heptapeptide, Aspartic Acid-Lysine-Glutamine-Histidine-Cysteine-Arginine-Phenylalanine is shown below. Each amino acid is denoted by a three letter code (Asp-Lys-Gln-His-Cys-Arg-Phe)..
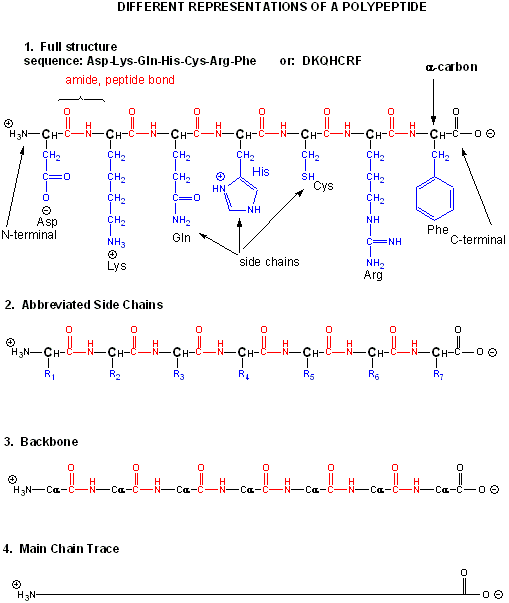
Notice that the protein chain has a beginning (an N-terminus with a amino group) and an end (a C-terminus with a carboxyl group). Also note that every atom in the backbone has a slight charge arising from the presence of the electronegative atoms O and N. Hence the backbone is polar. The R groups on each amino acid in the peptide are also called side chains.
The actual linear sequence of a protein is called its primary (1o) structure). Both the sequence of a protein and it's total length differ from one protein to another. Just for an octapeptide, there are over 25 billion different possible arrangement of amino acids. Hence the diversity of possible proteins is enormous.
Most proteins do not form an elongated structure as implied by the extended structures shown above. Rather they collapse on themselves to form compact, mostly globular (roughly spherical) structures. They do so as groups local and distant on the chain attract each other through IMFs which are now exerted within the large protein and not between different proteins. What kind of IMFs are involved?
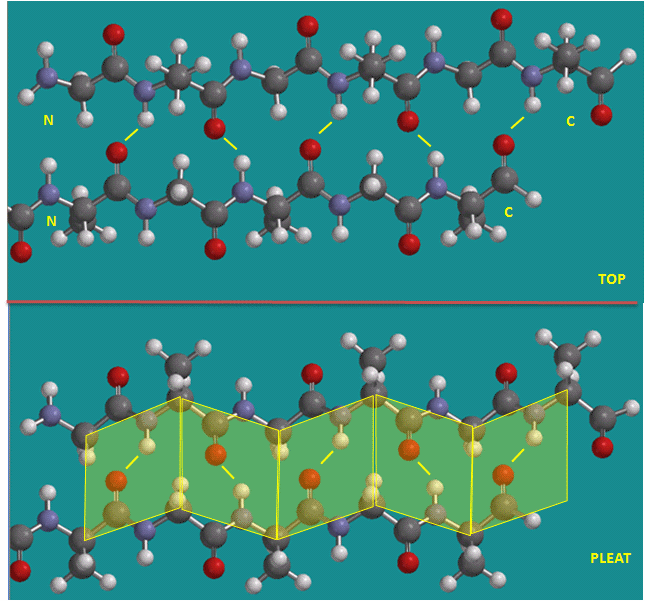
To simplify the process, lets consider first just the polar backbone without the side chains. The main chain can clearly form hydrogen bonds with itself and to water. If the hydrogen bonds are between the amide H (a hydrogen bond "donor" and a carbonyl O (a hydrogen bond "acceptor") a fixed number of amino acids distant from the amide H, a regular, repetitive secondary (2o) structures called a helix can form. One especially prevalent helix, the alpha helix, forms when within a short stretch of amino acids, the amide H of an amino acid in the backbone forms a hydrogen bond to the carbonyl C four amino acids in the protein sequence. Beta strands/sheets also occur when H bonds form between adjacent short stretches of amino acids in which the backbone of the short stretches are running either in the same N-C direction (parallel beta strands) or in opposite directions (antiparallel beta strands). The hydrogen bonds in secondary structures are all among main chain atoms in the backbone, not among side chains. The trace of an alpha helix in a protein is usually represented by a red or purple curly ribbon while beta traces are represented by yellow flat ribbons with an arrow showing the direction of the protein backbone from the N-terminus to C terminus direction.
The figures and Jmol models below illustrate secondary structure.
Alpha Helix (dotted yellow lines represent hydrogen bonds)
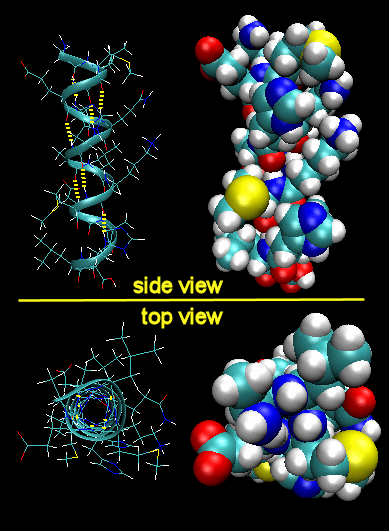
Note that all the side chains (R groups) are pointing away from the helix axis. Also evident from the space-filling model is that there is not opening in the helix as you look down the axis. The actual atoms are densely packed.
Antiparallel Beta Strands (yellow lines represent hydrogen bonds)

Parallel Beta Strands (yellow lines represent hydrogen bonds)
-
Jmol model of Alpha Helices - Observe the intrastrand H bond holding the helix together.
-
Jmol model of twisted parallel Beta Sheet - Notice the parallel natures of the strands. The protein sequences connecting these strands are removed for clarity
Protein folding is determined by much more than the formation of hydrogen bonds between backbone donors and acceptors. We must consider the effects of the 20 different R groups (side chains) which complicate the folding process. A protein ultimately folds in space to form a unique 3D shape, which usually contains some alpha helices and beta sheets. The overall 3 D structure is called the tertiary (3o) structure of the protein. The 3D structure of a protein determines the function of the protein as its shape and surface charge characteristics determines which molecules, both small and large, bind to the protein.
Here are some models of proteins showing secondary and tertiary structures.
- Jmol model of Myoglobin - an oxygen binding protein - Observe the predominately alpha-helical nature of the protein
- Jmol model of Superoxide Dismutase - a protein catalyst (enzyme) which breaks down superoxide, a toxic oxygen byproducts. High levels of this protein have been associated with longer life spans. Observe the predominate beta sheet structure of the protein.
- Jmol model of Triose Phosphate Isomerase - an enzyme involved in sugar metabolism. Notice the combination of alpha and beta secondary structure.
The structure of proteins is much more complicated than micelles and bilayers. To a first approximation the protein consists of a polar main chain/backbone from which amino acid side chains of varying polarity and charge hang. These side chains are polar uncharged, polar charged, and nonpolar. In general the nonpolar side chains are more stable buried in the center of the protein, surrounded by other nonpolar side chains and away from polar water. Compare this to the structure of a micelle. Given the greater complexity of protein primary and tertiary structure, however, not all nonpolar side groups can be buried. Some are on the surface exposed to solvent. Likewise, polar and charged polar side chains like to be on the surface exposed to water, but some will find themselves buried. If they are, they will be surrounded by polar side chains or interact with buried hydrogen bond donors and acceptors on the backbone that stabilize the buried polar group. Here are some findings about proteins derived from the known 3D structure of thousands of different proteins:
- On average, about 50% of the amino acids in a protein are in secondary structure. On average, there is about 27% alpha helix, and 23% beta structure.
- The side chain location varies with polarity. 83% of nonpolar side chains (such as Val, Leu, Ile, Met, and Phe) are in the interior in the folded protein.
- Charged polar side chains are almost equally partitioned between being buried or exposed on the surface.
- Uncharged polar groups such as Asn, Gln, Ser, Thr, and Tyr are mostly (63%) buried, and not on the surface;
- Globular (spherical) proteins are quite compact, with water excluded. The packing density (Vvdw/Vtot) is about 0.74, which is like the NaCl crystal and equals the closest packing density of 0.74. This compares to organic liquids, whose density is about 0.6-0.7.
- The packing around a buried nonpolar side chain of the amino acid phenylalanine (Phe) is shown in the Jmol below. It shows the structure of a small protein (protein tyrosine phosphatase) and the amino acids groups surrounding the buried Phe.
- Jmol model: Buried Phe in bovine protein tyrosine phosphatase
Contributors
Chris P Schaller, Ph.D., (College of Saint Benedict / Saint John's University)
{{template. Jakubowski()}}