
Statistical Entropy
- Page ID
- 96627
Entropy is a state function that is often erroneously referred to as the 'state of disorder' of a system. Qualitatively, entropy is simply a measure how much the energy of atoms and molecules become more spread out in a process and can be defined in terms of statistical probabilities of a system or in terms of the other thermodynamic quantities. Entropy is also the subject of the Second and Third laws of thermodynamics, which describe the changes in entropy of the universe with respect to the system and surroundings, and the entropy of substances, respectively.
Statistical Definition of Entropy
Entropy is a thermodynamic quantity that is generally used to describe the course of a process, that is, whether it is a spontaneous process and has a probability of occurring in a defined direction, or a non-spontaneous process and will not proceed in the defined direction, but in the reverse direction. To define entropy in a statistical manner, it helps to consider a simple system such as in Figure \(\PageIndex{1}\). Two atoms of hydrogen gas are contained in a volume of \( V_1 \).
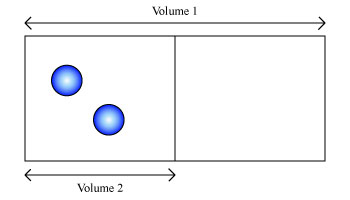
Since all the hydrogen atoms are contained within this volume, the probability of finding any one hydrogen atom in \( V_1 \) is 1. However, if we consider half the volume of this box and call it \( V_2 \),the probability of finding any one atom in this new volume is \( \frac{1}{2}\), since it could either be in \( V_2 \) or outside. If we consider the two atoms, finding both in \( V_2 \), using the multiplication rule of probabilities, is
\[ \frac{1}{2} \times \frac{1}{2} =\frac{1}{4}.\]
For finding four atoms in \( V_2 \) would be
\[ \frac{1}{2} \times \frac{1}{2} \times \frac{1}{2} \times \frac{1}{2}= \frac{1}{16}.\]
Therefore, the probability of finding N number of atoms in this volume is \( \frac{1}{2}^N \). Notice that the probability decreases as we increase the number of atoms.
If we started with volume \( V_2 \) and expanded the box to volume \( V_1 \), the atoms would eventually distribute themselves evenly because this is the most probable state. In this way, we can define our direction of spontaneous change from the lowest to the highest state of probability. Therefore, entropy \(S\) can be expressed as
\[ S=k_B \ln{\Omega} \label{1}\]
where \(\Omega\) is the probability and \( k_B \) is a proportionality constant. This makes sense because entropy is an extensive property and relies on the number of molecules, when \(\Omega\) increases to \( W^2 \), S should increase to 2S. Doubling the number of molecules doubles the entropy.
So far, we have been considering one system for which to calculate the entropy. If we have a process, however, we wish to calculate the change in entropy of that process from an initial state to a final state. If our initial state 1 is \( S_1=K_B \ln{\Omega}_1 \) and the final state 2 is \( S_2=K_B\ln{\Omega}_2 \),
\[ \Delta S=S_2-S_1=k_B \ln \dfrac{\Omega_2}{\Omega_1} \label{2}\]
using the rule for subtracting logarithms. However, we wish to define \(\Omega\) in terms of a measurable quantity. Considering the system of expanding a volume of gas molecules from above, we know that the probability is proportional to the volume raised to the number of atoms (or molecules), \( \alpha V^{N}\). Therefore,
\[ \Delta S=S_2-S_1=k_B \ln \left(\dfrac{V_2}{V_1} \right)^N=Nk_B \ln \dfrac{V_2}{V_1} \label{3} \]
We can define this in terms of moles of gas and not molecules by setting the \( k_{B} \) or Boltzmann constant equal to \( \frac{R}{N_A} \), where R is the gas constant and \( N_A \) is Avogadro's number. So for a expansion of an ideal gas and holding the temperature constant,
\[ \Delta S=\dfrac{N}{N_A} R \ln \left(\dfrac{V_2}{V_1} \right)^N=nR\ln \dfrac{V_2}{V_1} \label{4}\]
because \( \frac{N}{N_A}=n \), the number of moles. This is only defined for constant temperature because entropy can change with temperature. Furthermore, since S is a state function, we do not need to specify whether this process is reversible or irreversible.
Thermodynamic Definition of Entropy
Using the statistical definition of entropy is very helpful to visualize how processes occur. However, calculating probabilities like \(\Omega\) can be very difficult. Fortunately, entropy can also be derived from thermodynamic quantities that are easier to measure. Recalling the concept of work from the first law of thermodynamics, the heat (q) absorbed by an ideal gas in a reversible, isothermal expansion is
\[ q_{rev}=nRT\ln \dfrac{V_2}{V_1} \; . \label{5}\]
If we divide by T, we can obtain the same equation we derived above for \( \Delta S\):
\[ \Delta S=\dfrac{q_{rev}}{T}=nR\ln \dfrac{V_2}{V_1} \;. \label{6}\]
We must restrict this to a reversible process because entropy is a state function, however the heat absorbed is path dependent. An irreversible expansion would result in less heat being absorbed, but the entropy change would stay the same. Then, we are left with
\[ \Delta S> \dfrac{q_{irrev}}{T} \]
for an irreversible process because
\[ \Delta S=\Delta S_{rev}=\Delta S_{irrev} .\]
This apparent discrepancy in the entropy change between an irreversible and a reversible process becomes clear when considering the changes in entropy of the surrounding and system, as described in the second law of thermodynamics.
It is evident from our experience that ice melts, iron rusts, and gases mix together. However, the entropic quantity we have defined is very useful in defining whether a given reaction will occur. Remember that the rate of a reaction is independent of spontaneity. A reaction can be spontaneous but the rate so slow that we effectively will not see that reaction happen, such as diamond converting to graphite, which is a spontaneous process.
The Second Law as Energy Dispersion
Energy of all types -- in chemistry, most frequently the kinetic energy of molecules (but also including the phase change/potential energy of molecules in fusion and vaporization, as well as radiation) changes from being localized to becoming more dispersed in space if that energy is not constrained from doing so. The simplest example stereotypical is the expansion illustrated in Figure 1.
The initial motional/kinetic energy (and potential energy) of the molecules in the first bulb is unchanged in such an isothermal process, but it becomes more widely distributed in the final larger volume. Further, this concept of energy dispersal equally applies to heating a system: a spreading of molecular energy from the volume of greater-motional energy (“warmer”) molecules in the surroundings to include the additional volume of a system that initially had “cooler” molecules. It is not obvious, but true, that this distribution of energy in greater space is implicit in the Gibbs free energy equation and thus in chemical reactions.
“Entropy change is the measure of how more widely a specific quantity of molecular energy is dispersed in a process, whether isothermal gas expansion, gas or liquid mixing, reversible heating and phase change, or chemical reactions.” There are two requisites for entropy change.
- It is enabled by the above-described increased distribution of molecular energy.
- It is actualized if the process makes available a larger number of arrangements for the system’s energy, i.e., a final state that involves the most probable distribution of that energy under the new constraints.
Thus, “information probability” is only one of the two requisites for entropy change. Some current approaches regarding “information entropy” are either misleading or truly fallacious, if they do not include explicit statements about the essential inclusion of molecular kinetic energy in their treatment of chemical reactions.
References
- This literal greater spreading of molecular energy in 3-D space in an isothermal process is accompanied by occupancy of more quantum states (“energy levels”) within each microstate and thus more microstates for the final macrostate (i.e., a larger \(\Omega\) in R\ln{W}). Similarly, in any thermal process higher energy quantum states can be significantly occupied – thereby increasing the number of microstates in the product macrostate as measured by the Boltzmann relationship.
- I. N. Levine, Physical Chemistry, 6th ed. 2009, p. 101, toward the end of “What Is Entropy?”
- Chang, Raymond. Physical Chemistry for the Biosciences. Sausalito, California: University Science Books, 2005.
Contributors and Attributions
- Frank L. Lambert, Professor Emeritus, Occidental College
- Konstantin Malley (UCD)