
5.2: Molecular Structure- Theory and Experiment
- Page ID
- 11587

Experimental Probes of Molecular Shapes
I expect you are wondering why I want to discuss how experiments measure molecular shapes in this text whose aim is to introduce you to the field of theoretical chemistry. In fact, theory and experimental measurement are very connected, and it is these connections that I wish to emphasize in the following discussion. In particular, I want to make it clear that experimental data can only be interpreted, and thus used to extract molecular properties, through the application of theory. So, theory does not replace experiment, but serves both as a complementary component of chemical research (via. simulation of molecular properties) and as the means by which we connect laboratory data to molecular properties.
Rotational Spectroscopy
Most of us use rotational excitation of molecules in our every-day life. In particular, when we cook in a microwave oven, the microwave radiation, which has a frequency in the \(10^9- 10^{11} s^{-1}\) range, inputs energy into the rotational motions of the (primarily) water molecules contained in the food. These rotationally hot water molecules then collide with neighboring molecules (i.e., other water as well as proteins and other molecules in the food and in the cooking vessel) to transfer some of their motional energy to them. Through this means, the translational kinetic energy of all the molecules inside the cooker gains energy. This process of rotation-to-translation energy transfer is how the microwave radiation ultimately heats the food, which cooks it. What happens when you put the food into the microwave oven in a metal container or with some other metal material? As shown in Chapter 2, the electrons in metals exist in very delocalized partially filled orbitals called bands. These band orbitals are spread out throughout the entire piece of metal. The application of any external electric field (e.g., that belonging to the microwave radiation) causes these metal electrons to move throughout the metal. As these electrons accumulate more and more energy from the microwave radiation, they eventually have enough kinetic energy to be ejected into the surrounding air forming a discharge. This causes the sparking that we see when we make the mistake of putting anything metal into our microwave oven. Let’s now learn more about how the microwave photons cause the molecules to become rotationally excited.
Using microwave radiation, molecules having dipole moment vectors (\(\boldsymbol{\mu}\)) can be made to undergo rotational excitation. In such processes, the time-varying electric field \(\textbf{E} \cos(\omega t)\) of the microwave electromagnetic radiation interacts with the molecules via a potential energy of the form \(V = \textbf{E} \cdot \boldsymbol{\mu} \cos(\omega t)\). This potential can cause energy to flow from the microwave energy source into the molecule’s rotational motions when the energy of the former \(\hbar\omega/2\pi\) matches the energy spacing between two rotational energy levels.
This idea of matching the energy of the photons to the energy spacings of the molecule illustrates the concept of resonance and is something that is ubiquitous in spectroscopy as we learned in mathematical detail in Chapter 4. Upon first hearing that the photon’s energy must match an energy-level spacing in the molecule if photon absorption is to occur, it appears obvious and even trivial. However, upon further reflection, there is more to such resonance requirements than one might think. Allow me to illustrate using this microwave-induced rotational excitation example by asking you to consider why photons whose energies \(\hbar\omega/2\pi\) considerably exceed the energy spacing \(\Delta{E}\) will not be absorbed in this transition. That is, why is more than enough energy not good enough? The reason is that for two systems (in this case the photon’s electric field and the molecule’s rotation which causes its dipole moment to also rotate) to interact and thus exchange energy (this is what photon absorption is), they must have very nearly the same frequencies. If the photon’s frequency (\(\omega\)) exceeds the rotational frequency of the molecule by a significant amount, the molecule will experience an electric field that oscillates too quickly to induce a torque on the molecule's dipole that is always in the same direction and that lasts over a significant length of time. As a result, the rapidly oscillating electric field will not provide a coherent twisting of the dipole and hence will not induce rotational excitation.
One simple example from every day life can further illustrate this issue. When you try to push your friend, spouse, or child on a swing, you move your arms in resonance with the swinging person’s movement frequency. Each time the person returns to you, your arms are waiting to give a push in the direction that gives energy to the swinging individual. This happens over and over again; each time they return, your arms have returned to be ready to give another push in the same direction. In this case, we say that your arms move in resonance with the swing’s motion and offer a coherent excitation of the swinger. If you were to increase greatly the rate at which your arms are moving in their up and down pattern, the swinging person would not always experience a push in the correct direction when they return to meet your arms. Sometimes they would feel a strong in-phase push, but other times they would feel an out-of-phase push in the opposite direction. The net result is that, over a long period of time, they would feel random jerks from your arms, and thus would not undergo smooth energy transfer from you. This is why too high a frequency (and hence too high an energy) does not induce excitation. Let us now return to the case of rotational excitation by microwave photons.
As we saw in Chapter 2, for a rigid diatomic molecule, the rotational energy spacings are given by
\[E_{J+1} – E_J = 2 (J+1) \bigg(\dfrac{\hbar^2}{2I} \bigg) = 2\hbar c B (J+1) \tag{5.2.1}\]
where
\(I\) is the moment of inertia of the molecule given in terms of its equilibrium bond length \(r_e\) and its reduced mass \(\mu=\dfrac{m_am_b}{m_a+m_b}\) as \(I = Mr_e^2\). Thus, in principle, measuring the rotational energy level spacings via microwave spectroscopy allows one to determine \(r_e\). The second identity above simply defines what is called the rotational constant \(B\) in terms of the moment of inertia. The rotational energy levels described above give rise to a manifold of levels of non-uniform spacing as shown in the Figure 5.13.
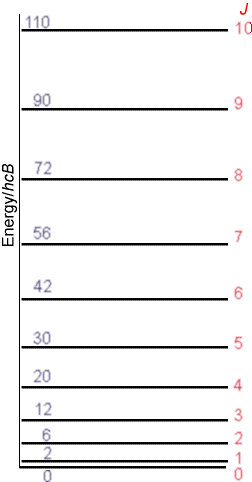
The non-uniformity in spacings is a result of the quadratic dependence of the rotational energy levels \(E_J\) on the rotational quantum number \(J\):
\[ E_J = J(J+1) \bigg(\dfrac{\hbar^2}{2I}\bigg).\tag{5.2.2}\]
Moreover, the level with quantum number \(J\) is \((2J+1)\)-fold degenerate; that is, there are \(2J+1\) distinct energy states and wave functions that have energy \(E_J\) and that are distinguished by a quantum number \(M\). These \(2J+1\) states have identical energy but differ among one another by the orientation of their angular momentum in space (i.e., the orientation of how they are spinning).
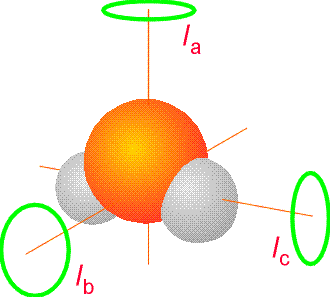
For polyatomic molecules, we know from Chapter 2 that things are more complicated because the rotational energy levels depend on three so-called principal moments of inertia (\(I_a\), \(I_b\), \(I_c\)) which, in turn, contain information about the molecule’s geometry. These three principle moments are found by forming a 3x3 moment of inertia matrix having elements
\[I_{x,x} = \sum_a m_a [ (R_a-R_{\rm CofM})^2 -(x_a - x_{\rm CofM} )^2\tag{5.2.3a}\]
and
\[I_{x,y} = \sum_a ma [ (x_a - x_{\rm CofM}) ( y_a -y_{\rm CofM}) ]\tag{5.2.3b}\]
expressed in terms of the Cartesian coordinates of the nuclei (a) and of the center of mass in an arbitrary molecule-fixed coordinate system (analogous definitions hold for \(I_{z,z}\), \(I_{y,y}\), \(I_{x,z}\) and \(I_{y,z}\)). The principle moments are then obtained as the eigenvalues of this 3x3 matrix.
For molecules with all three principle moments equal, the rotational energy levels are given by \(E_{J,K} = \dfrac{\hbar^2J(J+1)}{2I}\), and are independent of the \(K\) quantum number and on the \(M\) quantum number that again describes the orientation of how the molecule is spinning in space. Such molecules are called spherical tops. For molecules (called symmetric tops) with two principle moments equal (\(I_a\))) and one unique moment \(I_c\), the energies depend on two quantum numbers \(J\) and \(K\) and are given by
\[E_{J,K} = \dfrac{\hbar^2J(J+1)}{2I_a} + \hbar^2K^2 \bigg(\dfrac{1}{2I_c} - \dfrac{1}{2I_a}\bigg). \tag{5.2.4}\]
Species having all three principal moments of inertia unique, termed asymmetric tops, have rotational energy levels for which no analytic formula is yet known. The H2O molecule, shown in Figure 5.14, is such an asymmetric top molecule. More details about the rotational energies and wave functions were given in Chapter 2.
The moments of inertia that occur in the expressions for the rotational energy levels involve positions of atomic nuclei relative to the center of mass of the molecule. So, a microwave spectrum can, in principle, determine the moments of inertia and hence the geometry of a molecule. In the discussion given above, we treated these positions, and thus the moments of inertia as fixed (i.e., not varying with time). Of course, these distances are not unchanging with time in a real molecule because the molecule’s atomic nuclei undergo vibrational motions. Because of this, it is the vibrationally-averaged moments of inertia that must be incorporated into the rotational energy level formulas. Specifically, because the rotationally energies depend on the inverses of moments of inertia, one must vibrationally average \((R_a–R_{\rm CofM})^{-2}\) over the vibrational motion that characterizes the molecule’s movement. For species containing stiff bonds, the vibrational average \(\langle \psi|(R_a –R_{\rm CofM})^{-2}|\psi\rangle \) of the inverse squares of atomic distances relative to the center of mass does not differ significantly from the equilibrium values
\((R_{a,eq} –R_{\rm CofM})^{-2}\) of the same distances. However, for molecules such as weak van der Waals complexes (e.g., (H2O)2 or Ar..HCl) that undergo floppy large-amplitude vibrational motions, there may be large differences between the equilibrium \((R_{a,eq} –R_{\rm CofM})^{-2}\) and the vibrationally averaged values \(\langle \psi|(R_a –R_{\rm CofM})^{-2}|\psi\rangle \). The proper treatment of the rotational energy level patterns in such floppy molecules is still very much under active study by theoretical and experimental chemists. For this reason, it is a very challenging task to use microwave data on rotational energies to determine geometries (equilibrium or vibrationally averaged) for these kinds of molecules.
So, in the area of rotational spectroscopy theory plays several important roles:
- It provides the basic equations in terms of which the rotational line spacings relate to moments of inertia.
- It allows one, given the distribution of geometrical bond lengths and angles characteristic of the vibrational state the molecule exists in, to compute the proper vibrationally-averaged moment of inertia.
- It can be used to treat large amplitude floppy motions (e.g., by simulating the nuclear motions on a Born-Oppenheimer energy surface), thereby allowing rotationally resolved spectra of such species to provide proper moment of inertia (and thus geometry) information.
Vibrational Spectroscopy
The ability of molecules to absorb and emit infrared radiation as they undergo transitions among their vibrational energy levels is critical to our planet’s health. It turns out that water and \(CO_2\) molecules have bonds that vibrate in the \(10^{13}-10^{14} s^{-1}\) frequency range which is within the infrared spectrum (\(10^{11} –10^{14} s^{-1}\)). As solar radiation (primarily visible and ultraviolet) impacts the earth’s surface, it is absorbed by molecules with electronic transitions in this energy range (e.g., colored molecules such as those contained in plant leaves and other dark material). These molecules are thereby promoted to excited electronic states. Some such molecules re-emit the photons that excited them but most undergo so-called radiationless relaxation that allows them to return to their ground electronic state but with a substantial amount of internal vibrational energy. That is, these molecules become vibrationally very hot. Subsequently, these hot molecules, as they undergo transitions from high-energy vibrational levels to lower-energy levels, emit infrared (IR) photons.
If our atmosphere were devoid of water vapor and \(CO_2\), these IR photons would travel through the atmosphere and be lost into space. The result would be that much of the energy provided by the sun’s visible and ultraviolet photons would be lost via IR emission. However, the water vapor and \(CO_2\) do not allow so much IR radiation to escape. These greenhouse gases absorb the emitted IR photons to generate vibrationally hot water and \(CO_2\) molecules in the atmosphere. These vibrationally excited molecules undergo collisions with other molecules in the atmosphere and at the earth’s surface. In such collisions, some of their vibrational energy can be transferred to translational kinetic energy of the collision-partner molecules. In this manner, the temperature (which is measure of the average translational energy) increases. Of course, the vibrationally hot molecules can also re-emit their IR photons, but there is a thick layer of such molecules forming a blanket around the earth, and all of these molecules are available to continually absorb and re-emit the IR energy. In this manner, the blanket keeps the IR radiation from escaping and thus keeps our atmosphere warm. Those of us who live in dry desert climates are keenly aware of such effects. Clear cloudless nights in the desert can become very cold, primarily because much of the day’s IR energy production is lost to radiative emission through the atmosphere and into space. Let’s now learn more about molecular vibrations, how IR radiation excites them, and what theory has to do with this.
When infrared (IR) radiation is used to excite a molecule, it is the vibrations of the molecule that are in resonance with the oscillating electric field \(\textbf{E} \cos(\omega t)\). Molecules that have dipole moments that vary as its vibrations occur interact with the IR electric field via a potential energy of the form \(V = (\partial \boldsymbol{\mu}/\partial Q)\bullet\textbf{E} \cos(\omega t)\). Here \(\partial \boldsymbol{\mu}/\partial Q\) denotes the change in the molecule’s dipole moment \(M\) associated with motion along the vibrational normal mode labeled \(Q\).
As the IR radiation is scanned, it comes into resonance with various vibrations of the molecule under study, and radiation can be absorbed. Knowing the frequencies at which radiation is absorbed provides knowledge of the vibrational energy level spacings in the molecule. Absorptions associated with transitions from the lowest vibrational level to the first excited lever are called fundamental transitions. Those connecting the lowest level to the second excited state are called first overtone transitions. Excitations from excited levels to even higher levels are named hot-band absorptions.
Fundamental vibrational transitions occur at frequencies that characterize various functional groups in molecules (e.g., O-H stretching, H-N-H bending, N-H stretching, C-C stretching, etc.). As such, a vibrational spectrum offers an important fingerprint that allows the chemist to infer which functional groups are present in the molecule. However, when the molecule contains soft floppy vibrational modes, it is often more difficult to use information about the absorption frequency to extract quantitative information about the molecule’s energy surface and its bonding structure. As was the case for rotational levels of such floppy molecules, the accurate treatment of large-amplitude vibrational motions of such species remains an area of intense research interest within the theory community.
In a polyatomic molecule with \(N\) atoms, there are many vibrational modes. The total vibrational energy of such a molecule can be approximated as a sum of terms, one for each of the \(3N-6\) (or \(3N-5\) for a linear molecule) vibrations:
\[E(v_1 ... v_{3N-5\text{ or }6}) = \hbar\omega_j \big(v_j + \dfrac{1}{2}\big). \tag{5.2.1}\]
Here, \(\omega_j\) is the harmonic frequency of the \(j^{\rm th}\) mode and \(v_j\) is the vibrational quantum number associated with that mode. As we discussed in Chapter 3, the vibrational wave functions are products of harmonic vibrational functions for each mode:
\[\psi = \prod_{j=1}^{3N-5\text{ or }6} \psi_{v_j} (x (j)),\tag{5.2.1}\]
and the spacings between energy levels in which one of the normal-mode quantum numbers increases by unity are expressed as
\[\Delta E_{v_j} = E(...v_{j+1} ...) - E (...v_j ...) = \hbar\omega_j.\]
That is, the spacings between successive vibrational levels of a given mode are predicted to be independent of the quantum number v within this harmonic model as shown in Figure 5.15.
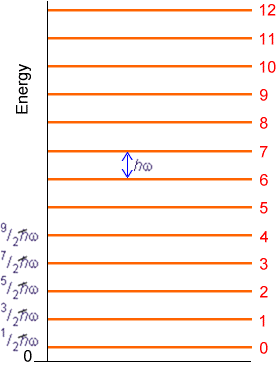
In Chapter 3, the details connecting the local curvature (i.e., Hessian matrix elements) in a polyatomic molecule’s potential energy surface to its normal modes of vibration are presented.
Experimental evidence clearly indicates that significant deviations from the harmonic oscillator energy expression occur as the quantum number \(v_j\) grows. These deviations are explained in terms of the molecule's true potential \(V(R)\) deviating strongly from the harmonic \(\dfrac{1}{2}k (R-R_e)^2\) potential at higher energy as shown in the Figure 5.16.
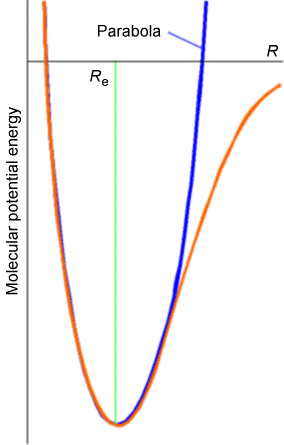
At larger bond lengths, the true potential is softer than the harmonic potential, and eventually reaches its asymptote, which lies at the dissociation energy \(D_e\) above its minimum. This deviation of the true \(V(R)\) from \(\dfrac{1}{2} k(R-R_e)^2\) causes the true vibrational energy levels to lie below the harmonic predictions.
It is convention to express the experimentally observed vibrational energy levels, along each of the \(3N-5\) or \(6\) independent modes in terms of an anharmonic formula similar to what we discussed for the Morse potential in Chapter 2:
\[E(v_j) = h\bigg[\omega_j \big(v_j + \dfrac{1}{2}\big) - (\omega_x)_j \big(v_j + \dfrac{1}{2}\big)^2 + (\omega_y)_j \big(v_j + \dfrac{1}{2}\big)^3 + (\omega_z)_j \big(v_j + \dfrac{1}{2}\big)^4 + ... \bigg]\]
The first term is the harmonic expression. The next is termed the first anharmonicity; it (usually) produces a negative contribution to \(E(v_j)\) that varies as \(\big(v_j + \dfrac{1}{2}\big)^2\). Subsequent terms are called higher anharmonicity corrections. The spacings between successive \(v_j \rightarrow v_j + 1\) energy levels are then given by:
\[\Delta{E}v_j = E(v_j + 1) - E(v_j)\tag{5.2.1}\]
\[= \hbar [\omega_j - 2(\omega_x)_j (v_j + 1) + ...]\tag{5.2.1}\]
A plot of the spacing between neighboring energy levels versus \(v_j\) should be linear for values of \(v_j\) where the harmonic and first anharmonicity terms dominate. The slope of
such a plot is expected to be \(-2\hbar(\omega_x)_j\) and the small \(-v_j\) intercept should be \(\hbar[\omega_j - 2(\omega_x)_j]\). Such a plot of experimental data, which clearly can be used to determine the \(\omega_j\) and \((\omega_x)_j\) parameters of the vibrational mode of study, is shown in Figure 5.17.
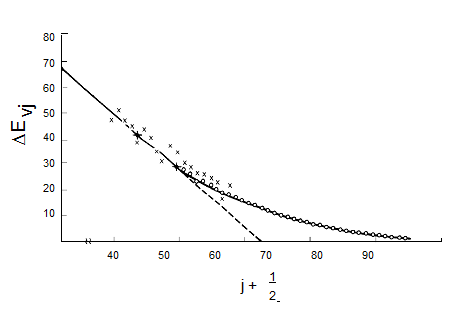
Figure 5.17 Birge-Sponer plot of vibrational energy spacings vs. quantum number.
These so-called Birge-Sponer plots can also be used to determine dissociation energies of molecules if the vibration whose spacings are plotted corresponds to a bond-stretching mode. By linearly extrapolating such a plot of experimental \(\Delta E_{v_j}\) values to large \(v_j\) values, one can find the value of \(v_j\) at which the spacing between neighboring vibrational levels goes to zero. This value \(v_j\), max specifies the quantum number of the last bound vibrational level for the particular bond-stretching mode of interest. The dissociation energy \(D_e\) can then be computed by adding to \(\dfrac{1}{2}\hbar\omega_j\) (the zero point energy along this mode) the sum of the spacings between neighboring vibrational energy levels from \(v_j = 0\) to \(v_j = v_{j, max}\):
\[D_e = \dfrac{1}{2}\hbar\omega_j + \sum_{v_j=0}^{v_{j,max}}\Delta D_{v_j}.\]
So, in the case of vibrational spectroscopy, theory allows us to
- interpret observed infrared lines in terms of absorptions arising in localized functional groups;
- extract dissociation energies if a long progression of lines is observed in a bond-stretching transition;
- and treat highly non-harmonic floppy vibrations by carrying out dynamical simulations on a Born-Oppenheimer energy surface.
X-Ray Crystallography
In x-ray crystallography experiments, one employs crystalline samples of the molecules of interest and makes use of the diffraction patterns produced by scattered x-rays to determine positions of the atoms in the molecule relative to one another using the famous Bragg formula:
\[n\lambda = 2d \sin \theta .\tag{5.2.1}\]
In this equation, \(\lambda\) is the wavelength of the x-rays, \(d\) is a spacing between layers (planes) of atoms in the crystal, \(q\) is the angle through which the x-ray beam is scattered, and \(n\) is an integer (1,2, …) that labels the order of the scattered beam.
Because the x-rays scatter most strongly from the inner-shell electrons of each atom, the interatomic distances obtained from such diffraction experiments are, more precisely, measures of distances between high electron densities in the neighborhoods of various atoms. The x-rays interact most strongly with the inner-shell electrons because it is these electrons whose characteristic Bohr frequencies of motion are (nearly) in resonance with the high frequency of such radiation. For this reason, x-rays can be viewed as being scattered from the core electrons that reside near the nuclear centers within a molecule. Hence, x-ray diffraction data offers a very precise and reliable way to probe inter-atomic distances in molecules.
The primary difficulties with x-ray measurements are:
- That one needs to have crystalline samples (often, materials simply cannot be grown as crystals),
- That one learns about inter-atomic spacings as they occur in the crystalline state, not as they exist, for example, in solution or in gas-phase samples. This is especially problematic for biological systems where one would like to know the structure of the bio-molecule as it exists within the living organism.
Nevertheless, x-ray diffraction data and its interpretation through the Bragg formula provide one of the most widely used and reliable ways for probing molecular structure.
NMR Spectroscopy
NMR spectroscopy probes the absorption of radio-frequency (RF) radiation by the nuclear spins of the molecule. The most commonly occurring spins in natural samples are \(^1H\) (protons), \(^2H\) (deuterons), \(^{13}C\) and \(^{15}N\) nuclei. In the presence of an external magnetic field \(B_{0z}\) along the \(z\)-axis, each such nucleus has its spin states split in energy by an amount given by \(B_0(1-\sigma_k)\gamma_k M_I\), where \(M_I\) is the component of the \(k^{\rm th}\) nucleus’ spin angular momentum along the \(z\)-axis, \(B_0\) is the strength of the external magnetic field, and \(\gamma_k\) is a so-called gyromagnetic factor (i.e., a constant) that is characteristic of the \(k^{\rm th}\) nucleus. This splitting of magnetic spin levels by a magnetic field is called the Zeeman effect, and it is illustrated in Figure 5.18.
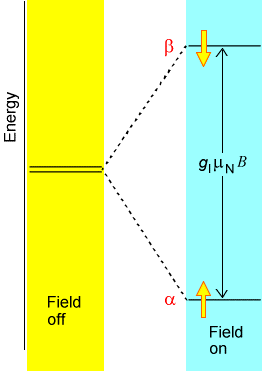
The factor \((1-\sigma_k)\) is introduced to describe the screening of the external \(B_0\)-field at the \(k^{\rm th}\) nucleus caused by the electron cloud that surrounds this nucleus. In effect, \(B_0(1-\sigma_k)\) is the magnetic field experienced local to the \(k^{\rm th}\) nucleus. It is this \((1-\sigma_k)\) screening that gives rise to the phenomenon of chemical shifts in NMR spectroscopy, and it is this factor that allows NMR measurements of shielding factors (\(\sigma_k\)) to be related, by theory, to the electronic environment of a nucleus. In Figure 5.19 we display the chemical shifts of proton and \(^{13}C\) nuclei in a variety of chemical bonding environments.
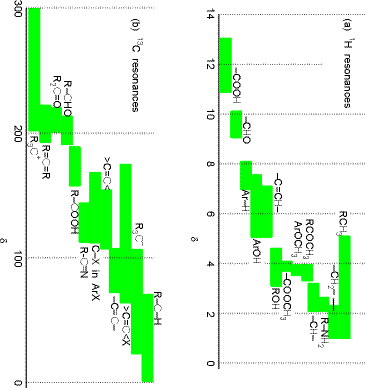
Because the \(M_I\) quantum number changes in steps of unity and because each photon possesses one unit of angular momentum, the RF energy \(\hbar\omega\) that will be in resonance with the \(k^{\rm th}\) nucleus’ Zeeman-split levels is given by \(\hbar\omega = B_0(1-\sigma_k)\gamma_k\).
In most NMR experiments, a fixed RF frequency is employed and the external magnetic field is scanned until the above resonance condition is met. Determining at what \(B_0\) value a given nucleus absorbs RF radiation allows one to determine the local shielding \((1-\sigma_k)\) for that nucleus. This, in turn, provides information about the electronic environment local to that nucleus as illustrated in the above figure. This data tells the chemist a great deal about the molecule’s structure because it suggests what kinds of functional groups occur within the molecule.
To extract even more geometrical information from NMR experiments, one makes use of another feature of nuclear spin states. In particular, it is known that the energy levels of a given nucleus (e.g., the \(k^{\rm th}\) one) are altered by the presence of other nearby nuclear spins. These spin-spin coupling interactions give rise to splittings in the energy levels of the \(k^{\rm th}\) nucleus that alter the above energy expression as follows:
\[E_M = B_0(1-\sigma_k)\gamma_k M+J_{M \, M’}\tag{5.2.1}\]
Where \(M\) is the \(z\)-component of the \(k^{\rm th}\) nuclear spin angular momentum, \(M’\) is the corresponding component of a nearby nucleus causing the splitting, and \(J\) is called the spin-spin coupling constant between the two nuclei.
Examples of how spins on neighboring centers split the NMR absorption lines of a given nucleus are shown in Figs. 5.20-5.22 for three common cases. The first involves a nucleus (labeled A) that is close enough to one other magnetically active nucleus (labeled X); the second involves a nucleus (A) that is close to two equivalent nuclei (2X); and the third describes a nucleus (A) close to three equivalent nuclei (X3).
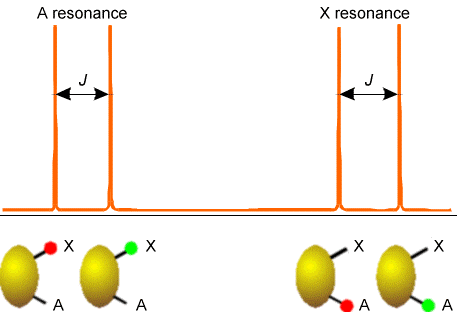
In Figure 5.20 are illustrated the splitting in the X nucleus’ absorption due to the presence of a single A neighbor nucleus (right) and the splitting in the A nucleus’ absorption (left) caused by the X nucleus. In both of these examples, the X and A nuclei have only two \(M_I\) values, so they must be spin-1/2 nuclei. This kind of splitting pattern would, for example, arise for a \(^{13}C-H\) group in the benzene molecule where A = \(^{13}C\) and X = \(^1H\).
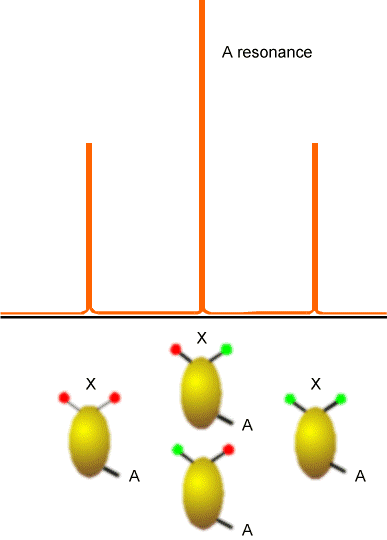
The (\(AX_2\)) splitting pattern shown if Figure 5.21 would, for example, arise in the \(^{13}C\) spectrum of a \(–CH_2^-\) group, and illustrates the splitting of the A nucleus’ absorption line by the four spin states that the two equivalent X spins can occupy. Again, the lines shown would be consistent with X and A both having spin 1/2 because they each assume only two \(M_I\) values.
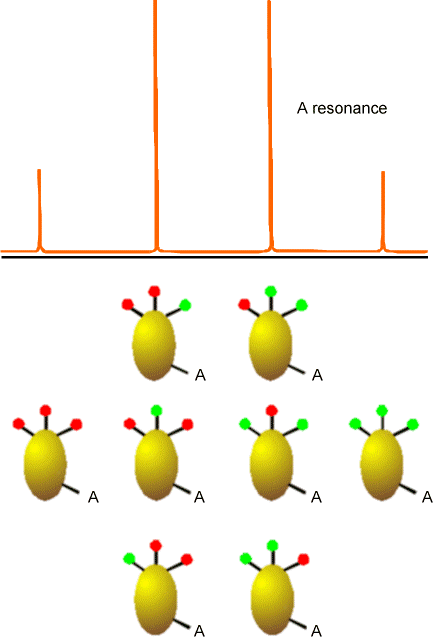
In Figure 5.22 is the kind of splitting pattern (\(AX_3\)) that would apply to the \(^{13}C\) NMR absorptions for a \(–CH_3\) group. In this case, the spin-1/2 A line is split by the eight spin states that the three equivalent spin-1/2 H nuclei can occupy.
The magnitudes of these \(J\) coupling constants depend on the distances R between the two nuclei to the inverse sixth power (i.e., as \(R^{-6}\)). They also depend on the \(g\) values of the two interacting nuclei. In the presence of splitting caused by nearby (usually covalently bonded) nuclei, the NMR spectrum of a molecule consists of sets of absorptions (each belonging to a specific nuclear type in a particular chemical environment and thus have a specific chemical shift) that are split by their couplings to the other nuclei. Because of the spin-spin coupling’s strong decay with internuclear distance, the magnitude and pattern of the splitting induced on one nucleus by its neighbors provides a clear signature of what the neighboring nuclei are (i.e., through the number of \(M’\) values associated with the peak pattern) and how far these nuclei are (through the magnitude of the \(J\) constant, knowing it is proportional to \(R^{-6}\)). This near-neighbor data, combined with the chemical shift functional group data, offer powerful information about molecular structure.
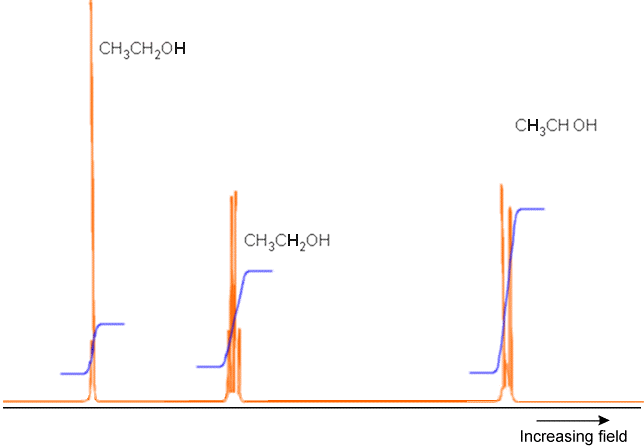
An example of a full NMR spectrum is given in Figure 5.23 where the \(^1H\) spectrum (i.e., only the proton absorptions are shown) of \(H_3C-H_2C-OH\) appears along with plots of the integrated intensities under each set of peaks. The latter data suggests the total number of nuclei corresponding to that group of peaks. Notice how the \(OH\) proton’s absorption, the absorption of the two equivalent protons on the \(–CH_2^-\) group, and that of the three equivalent protons in the \(–CH_3\) group occur at different field strengths (i.e., have different chemical shifts). Also note how the \(OH\) peak is split only slightly because this proton is distant from any others, but the \(CH_3\) protons peak is split by the neighboring \(–CH_2^-\) group’s protons in an \(AX_2\) pattern. Finally, the \(–CH_2^-\) protons’ peak is split by the neighboring \(–CH_3\) group’s three protons (in an \(AX_3\) pattern).
In summary, NMR spectroscopy is a very powerful tool that:
- allows us to extract inter-nuclear distances (or at least tell how many near-neighbor nuclei there are) and thus geometrical information by measuring coupling constants \(J\) and subsequently using the theoretical expressions that relate \(J\) values to \(R^{-6}\) values.
- allows us to probe the local electronic environment of nuclei inside molecules by measuring chemical shifts or shielding \(\sigma_I\) and then using the theoretical equations relating the two quantities. Knowledge about the electronic environment tells one, for example, about the degree of polarity in bonds connected to that nuclear center.
- tells us, through the splitting patterns associated with various nuclei, the number and nature of the neighbor nuclei, again providing a wealth of molecular structure information.
Theoretical Simulation of Structures
We have seen how microwave, infrared, and NMR spectroscopy as well as x-ray diffraction data, when subjected to proper interpretation using the appropriate theoretical equations, can be used to obtain a great deal of structural information about a molecule. As discussed in Part 1 of this text, theory is also used to probe molecular structure in another manner. That is, not only does theory offer the equations that connect the experimental data to the molecular properties, but it also allows one to simulate a molecule. This simulation is done by solving the Schrödinger equation for the motions of the electrons to generate a potential energy surface \(E(R)\), after which this energy landscape can be searched for points where the gradients along all directions vanish. An example of such a PES is shown in Figure 5.24 for a simple case in which the energy depends on only two geometrical parameters. Even in such a case, one can find several local minima and transition state structures connecting them.
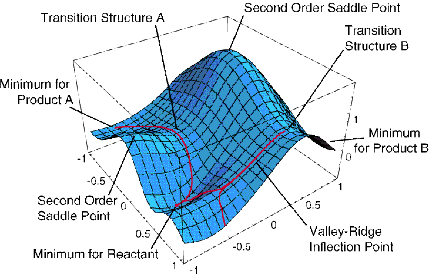
As we discussed in Chapter 3, among the stationary points on the potential energy surface (PES), those at which all eigenvalues of the second derivative (Hessian) matrix are positive represent geometrically stable isomers of the molecule. Those stationary points on the PES at which all but one Hessian eigenvalue are positive and one is negative represent transition state structures that connect pairs of stable isomers.
Once the stable isomers of a molecule lying within some energy interval above the lowest such isomer have been identified, the vibrational motions of the molecule within the neighborhood of each such isomer can be described either by solving the Schrödinger equation for the vibrational wave functions \(\chi_v(Q)\) belonging to each normal mode or by solving the classical Newton equations of motion using the gradient \(\dfrac{∂E}{∂Q}\) of the PES to compute the forces along each molecular distortion direction \(Q\):
\[F_Q = -\dfrac{\partial E}{\partial Q} \tag{5.2.1}\]
The decision about whether to use the Schrödinger or Newtonian equations to treat the vibrational motion depends on whether one wishes (needs) to properly include quantum effects (e.g., zero-point motion and wave function nodal patterns) in the simulation.
Once the vibrational motions have been described for a particular isomer, and given knowledge of the geometry of that isomer, one can evaluate the moments of inertia, one can properly vibrationally average all of the \(R^{-2}\) quantities that enter into these moments, and, hence, one can simulate the microwave spectrum of the molecule. Also, given the Hessian matrix for this isomer, one can form its mass-weighted variant whose non-zero eigenvalues give the normal-mode harmonic frequencies of vibration of that isomer and whose eigenvectors describe the atomic motions that correspond to these vibrations. Moreover, the solution of the electronic Schrödinger equation allows one to compute the NMR shielding \(\sigma_I\) values at each nucleus as well as the spin-spin coupling constants \(J\) between pairs of nuclei (the treatment of these subjects is beyond the level of this text; you can find it in Molecular Electronic Structure Theory by Helgaker, et. al.). Again, using the vibrational motion knowledge, one can average the \(\sigma\) and \(J\) values over this motion to gain vibrationally averaged \(\sigma_I\) and \(J_{I,I’}\) values that best simulate the experimental parameters.
One carries out such a theoretical simulation of a molecule for various reasons.
Especially in the early days of developing theoretical tools to solve the electronic Schrödinger equation or the vibrational motion problem, one would do so for molecules whose structures and IR and NMR spectra were well known. The purpose in such cases was to calibrate the accuracy of the theoretical methods against established experimental data. Now that theoretical tools have been reasonably well tested and can be trusted (within known limits of accuracy), one often uses theoretically simulated structural and spectroscopic properties to identify spectral features whose molecular origin is not known. That is, one compares the theoretical spectra of a variety of test molecules to the observed spectral features to attempt to identify the molecule that produced the spectra.
It is also common to use simulations to examine species that are especially difficult to generate in reasonable quantities in the laboratory and species that do not persist for long times. Reactive radicals, cations and anions are often difficult to generate in the laboratory and may be impossible to retain in sufficient concentrations and for a sufficient duration to permit experimental characterization. In such cases, theoretical simulation of the properties of these molecules may be the most reliable way to access such data. Moreover, one might use simulations to examine the behavior of molecules under extreme conditions such as high pressure, confinement to nanoscopic spaces, high temperature, or very low temperatures for which experiments could be very difficult or expensive to carry out.
Let me tell you about an example of how such theoretical simulation has proven useful, probably even essential, for interpreting experimental data (the data is reported in N. I. Hammer, J-W. Shin, J. M. Headrick, E. G. Diken, J. R. Roscioli, G. H. Weddle, and M. A. Johnson, Science 306, 675 (2004)). In the group of Prof. Mark Johnson at Yale, infrared spectroscopy is carried out on gas-phase ions. In this particular experiment, water cluster anions \(Ar_k(H_2O)_n^-\) with one or more Ar atoms attached to them were formed and, using a mass spectrometer, the ions of one specific mass were selected for subsequent study. In the example illustrated here, the cluster \(Ar_k(H_2O)_4^-\) containing four water molecules was studied.
When infrared (IR) radiation impinges on the \(Ar_k(H_2O)_4^-\) ions, it can be absorbed if its frequency matches the frequency of one of the vibrational modes of this cluster. If, for example, IR radiation in the 1500-1700 cm-1 frequency range is absorbed (this range corresponds to frequencies of H-O-H bending vibrations), this excess internal energy can cause one or more of the weakly bound Ar atoms to be ejected from the \(Ar_k(H_2O)_4^-\) cluster, thus decreasing the number of intact \(Ar_k(H_2O)_4^-\) ions in the mass spectrometer. The decrease in the number of intact ions is a direct measure then of the absorption of the IR light. By monitoring the number of \(Ar_k(H_2O)_4^-\) (i.e., the strength of the mass spectrometer’s signal at this particular mass-to-charge ratio) as the IR radiation is tuned through the 1500-1700 cm-1 frequency range, the experimentalists obtain spectral signatures (i.e., the ion intensity loss) of the IR absorption by the \(Ar_k(H_2O)_4^-\) cluster ions.
When they carry out this kind of experiment using \(Ar_5(H_2O)_4^-\) and scan the IR radiation in the 1500-1700 cm-1 frequency range, they obtained the spectrum labeled A in Figure 5. 24 a. When they performed the same kind of experiment on \(Ar_{10}(D_2O)_4^-\) and scanned in the 2400-2800 cm-1 frequency range (which is where O-D stretching vibrations are known to occur), they obtained the spectrum labeled B in Figure 5. 24 a.
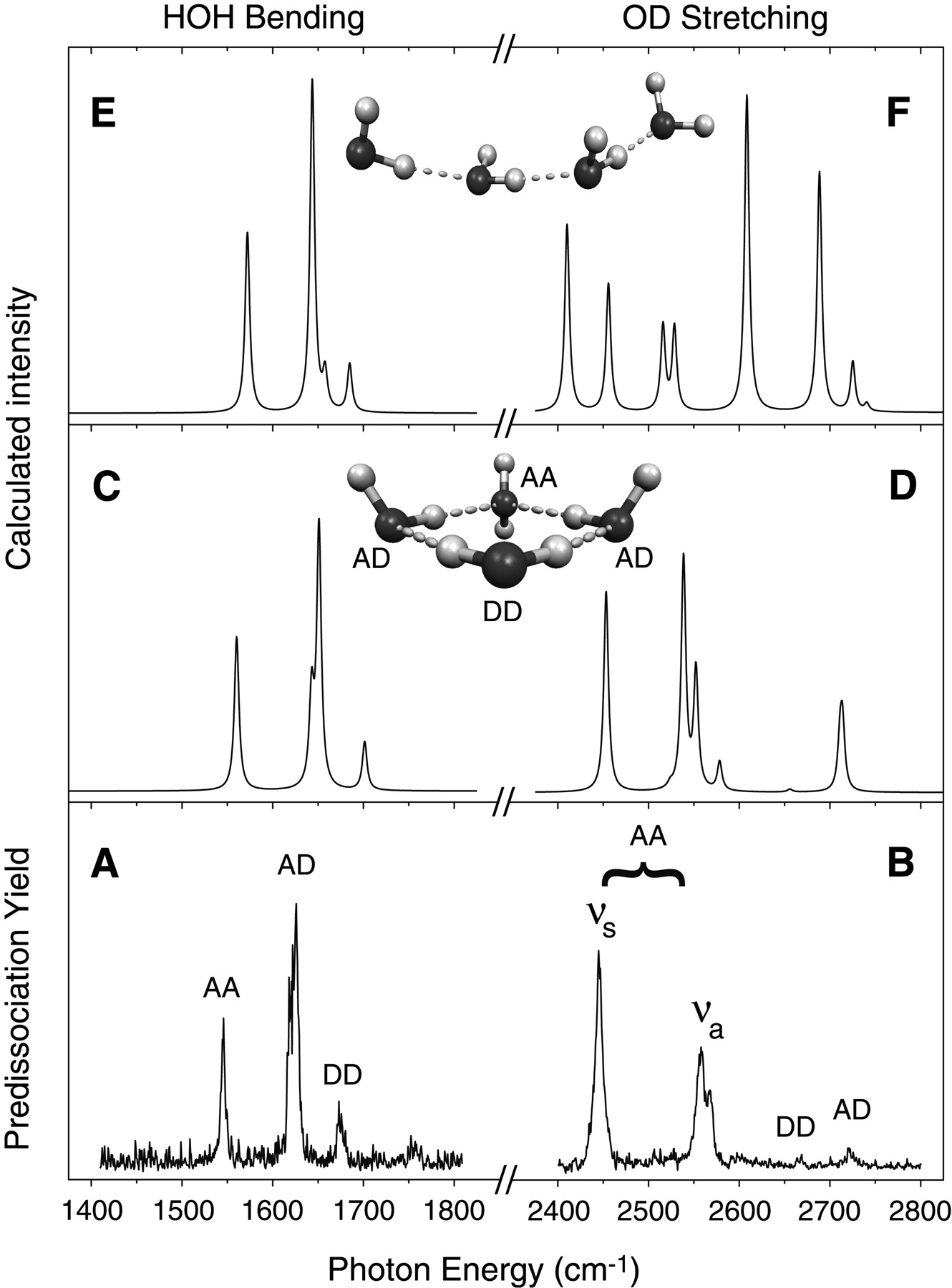
What the experimentalists did not know, however, is what the geometrical structure of the underlying \((H_2O)_4^-\) ion was. Nor did they know exactly which H-O-H bending or O-H (or O-D) stretching vibrations were causing the various peaks shown in Figure 5.24 a A and B.
By carrying out electronic structure calculations on a large number of geometries for \((H_2O)_4^-\) and searching for local minima on the ground electronic state of this ion (there are a very large number of such local minima) and then using the mass-weighted Hessian matrix at each local minima to calculate the structure’s vibrational energies, the experimentalists were able to figure out what structure for \((H_2O)_4^-\) was most consistent with their observed IR spectrum. For example, for the rather extended structure of \((H_2O)_4\), they computed the IR spectrum shown in panel E (and for \((D_2O)_4^-\) in panel F) of Figure 5. 24 a. Alternatively, for the cyclic structure of \((H_2O)_4\)-, they computed the IR spectrum shown in panel C (and for \((D_2O)_4^-\) in panel D) of Figure 5. 24 a. Clearly, the spectrum of panels C and D agrees much better with their experimental spectrum in panels A and B than does the spectrum of panels E and F. Based on these comparisons, these scientists concluded that the \((H_2O)_4^-\) ions in their \(Ar_5(H_2O)_4^-\) and \(Ar_{10}(D_2O)_4^-\) have the cyclic geometry, not the extended quasi-linear geometry. Moreover, by looking at which particular vibrational modes of the cyclic \((H_2O)_4^-\) produced which peaks in panels C and D, they were able to assign each of the IR peaks seen in their data of panels A and B. This is a good example of how theoretical simulation can help interpret experimental data; without the theory, these scientists would not know the geometry of \((H_2O)_4^-\).
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)