
4.1: Perturbation Theory
- Page ID
- 11580

In most practical applications of quantum mechanics to molecular problems, one is faced with the harsh reality that the Schrödinger equation pertinent to the problem at hand cannot be solved exactly. To illustrate how desperate this situation is, I note that neither of the following two Schrödinger equations has ever been solved exactly (meaning analytically):
- The Schrödinger equation for the two electrons moving about the He nucleus:
\[\left[- \dfrac{\hbar^2}{2m_e} \nabla_1^2 - \dfrac{\hbar^2}{2m_e} \nabla_2^2 – \frac{2e^2}{r_1} – \frac{2e^2}{r_2} + \frac{e^2}{r_{1,2}}\right] \psi= E \psi,\]
- The Schrödinger equation for the two electrons moving in an \(H_2\) molecule even if the locations of the two nuclei (labeled A and B) are held clamped as in the Born-Oppenheimer approximation:
\[\left[- \dfrac{\hbar^2}{2m_e} \nabla_1^2 - \dfrac{\hbar^2}{2m_e} \nabla_2^2 – \dfrac{e^2}{r_{1,A}} – \dfrac{e^2}{r_{2,A}} – \dfrac{e^2}{r_{1,B}} – \dfrac{e^2}{r_{2,B}} + \dfrac{e^2}{r_{1,2}}\right] \psi = E \psi\]
These two problems are examples of what is called the “three-body problem” meaning solving for the behavior of three bodies moving relative to one another. Motions of the sun, earth, and moon (even neglecting all the other planets and their moons) constitute another three-body problem. None of these problems, even the classical Newton’s equation for the sun, earth, and moon, have ever been solved exactly. So, what does one do when faced with trying to study real molecules using quantum mechanics?
There are two very powerful tools that one can use to “sneak up” on the solutions to the desired equations by first solving an easier model problem and then using the solutions to this problem to approximate the solutions to the real Schrödinger problem of interest. For example, to solve for the energies and wave functions of a boron atom, one could use hydrogenic \(1s\) orbitals (but with \(Z = 5\)) and hydrogenic \(2s\) and \(2p\) orbitals (with \(Z = 3\) to account for the screening of the full nuclear charge by the two \(1s\) electrons) as a starting point. To solve for the vibrational energies of a diatomic molecule whose energy vs. bond length \(E(R)\) is known, one could use the Morse oscillator wave functions and energies as starting points. But, once one has decided on a reasonable model to use, how does one connect this model to the real system of interest? Perturbation theory and the variational method are the two tools that are most commonly used for this purpose, and it is these two tools that are covered in this Chapter.
The perturbation theory approach provides a set of analytical expressions for generating a sequence of approximations to the true energy \(E\) and true wave function \(\psi\). This set of equations is generated, for the most commonly employed perturbation method, Rayleigh-Schrödinger perturbation theory (RSPT), as follows. First, one decomposes the true Hamiltonian \(H\) into a so-called zeroth-order part \(H^{(0)}\) (this is the Hamiltonian of the model problem used to represent the real system) and the difference (\(H-H^{(0)}\)), which is called the perturbation and usually denoted \(V\):
\[H = H^{(0)} + V.\]
It is common to associate with the perturbation \(V\) a strength parameter \(\lambda\), which could, for example, be associated with the strength of the electric field when the perturbation results from the interaction of the molecule of interest with an electric field. In such cases, it is usual to write the decomposition of \(H\) as
\[H = H^{(0)} + \lambda V\]
A fundamental assumption of perturbation theory is that the wave functions and energies for the full Hamiltonian \(H\) can be expanded in a Taylor series involving various powers of the perturbation parameter \(\lambda\). Hence, one writes the energy \(E\) and the wave function \(\psi\) as zeroth-, first-, second, etc, order pieces which form the unknowns in this method:
\[E = E^{(0)} + E^{(1)} + E^{(2)} + E^{(3)} + \cdots\]
\[y = \psi^{(0)} + \psi^{(1)} + \psi^{(2)} + \psi^{(3)} + \cdots\]
with \(E^{(n)}\) and \(\psi^{(n)}\) being proportional to \(\lambda_n\). Next, one substitutes these expansions of \(E\), \(H\) and \(\psi\) into \(H\psi = E\psi\). This produces one equation whose right and left hand sides both contain terms of various “powers” in the perturbation \(\lambda\). For example, terms of the form \(E^{(1)}\), \(\psi^{(2)}\), and \(V\) \(\psi^{(2)}\) and \(E^{(0)}\) \(\psi^{(3)}\) are all of third power (also called third order). Next, one equates the terms on the left and right sides that are of the same order. This produces a set of equations, each containing all the terms of a given order. The zeroth, first, and second-order such equations are given below:
\[H^{(0)} \psi^{(0)} = E^{(0)} \psi^{(0)},\]
\[H^{(0)} \psi^{(1)} + V \psi^{(0)} = E^{(0)} \psi^{(1)} + E^{(1)} \psi^{(0)}\]
\[H^{(0)} \psi^{(2)} + V \psi^{(1)} = E^{(0)} \psi^{(2)} + E^{(1)} \psi^{(1)} + E^{(2)} \psi^{(0)}.\]
It is straightforward to see that the nth order expression in this sequence of equations can be written as
\[H^{(0)} \psi^{(n)} + V \psi^{(n-1)} = E^{(0)} \psi^{(n)} + E^{(1)} \psi^{(n-1)} + E^{(2)} \psi^{(n-2)} + E^{(3)} \psi^{(n-3)} + \cdots + E^{(n)} \psi^{(0)}.\]
The zeroth-order equation simply instructs us to solve the model Schrödinger equation to obtain the zeroth-order wave function \(\psi^{(0)}\) and its zeroth-order energy \(E^{(0)}\). Since \(H^{(0)}\) is a Hermitian operator, it has a complete set of such eigenfunctions, which we label \(\{\psi^{(0)}k\}\) and {E^{(0)}_k}. One of these states will be the one we are interested in studying (e.g., we might be interested in the effect of an external electric field on the \(2s\) state of the hydrogen atom), but, as will become clear soon, we actually have to find the full set of {\(\psi^{(0)}_k\)} and {\(E^{(0)}_k\)} (e.g., we need to also find the \(1s, 2p, 3s, 3p, 3d,\) etc. states of the hydrogen atom when studying the electric field’s effect on the \(2s\) state).
In the first-order equation, the unknowns are \(\psi^{(1)}\) and \(E^{(1)}\) (recall that \(V\) is assumed to be known because it is the difference between the Hamiltonian one wants to solve and the model Hamiltonian \(H^{(0)}\)). To solve the first-order and higher-order equations, one expands each of the corrections to the wave function \(\psi\) of interest in terms of the complete set of wave functions of the zeroth-order problem \(\{\psi^{(0)}_J\}\). As noted earlier, this means that one must solve \(H^{(0)} \psi^{(0)}_J = E^{(0)}J \psi^{(0)}_J\) not just for the zeroth-order state one is interested in (denoted \(\psi^{(0)}\) above), but for all of the other zeroth-order states \(\{\psi^{(0)}_J\}\). For example, expanding \(\psi^{(1)}\) in this manner gives:
\[\psi^{(1)}=\sum_J C_J^1\psi_J^{(0)}\]
Now, the unknowns in the first-order equation become \(E^{(1)}\) and the expansion coefficients. To solve
\[H^{(0)} \psi^{(1)} + V \psi^{(0)} = E^{(0)} \psi^{(1)} + E^{(1)} \psi^{(0)}\]
one proceeds as follows:
1. First, one multiplies this equation on the left by the complex conjugate of the zeroth-order function for the state of interest \(\psi^{(0)}\) and integrates over the variables on which the wave functions depend. This gives
\[\langle\psi^{(0)}|H^{(0)}|\psi^{(1)}\rangle + \langle\psi^{(0)}|V|\psi^{(0)}\rangle = E^{(0)} \langle\psi^{(0)}|\psi^{(1)}\rangle + E^{(1)} \langle\psi^{(0)}|\psi^{(0)}\rangle.\]
The first and third terms cancel one another because \(H^{(0)} \psi^{(0)} = E^{(0)} \psi^{(0)}\), and the fourth term reduces to \(E^{(1)}\) because \(\psi^{(0)}\) is assumed to be normalized. This allows the above equation to be written as
\[E^{(1)} = \langle\psi^{(0)} | V | \psi^{(0)}\rangle\]
which is the RSPT expression for \(E^{(1)}\). It says the first-order correction to the energy \(E^{(0)}\) of the unperturbed state can be evaluated by computing the average value of the perturbation with respect to the unperturbed wave function \(\psi^{(0)}\).
2. Returning to the first-order equation and multiplying on the left by the complex conjugate of one of the other zeroth-order functions gives
\[\langle \psi_J^{(0)}|H^{(0)}|\psi^{(1)}\rangle + \langle\psi_J^{(0)} |V|\psi^{(0)}\rangle = E^{(0)} \langle\psi_J^{(0)}|\psi^{(1)}\rangle + E^{(1)} \langle \psi_J^{(0)}|\psi^{(0)}\rangle.\]
Using \(H^{(0)} = \), the first term reduces to \(\langle |\psi^{(1)}\rangle\), and the fourth term vanishes because is orthogonal to \(\psi^{(0)}\) because these two functions are different eigenfunctions of \(H^{(0)}\). This reduces the equation to
\[\langle \psi_J^{(0)}|\psi^{(1)}\rangle + \langle\psi_J^{(0)} |V|\psi^{(0)}\rangle = E^{(0)} \langle\psi_J^{(0)} |\psi^{(1)}\rangle\]
The unknown in this expression is \(\langle\psi_J^{(0)} |\psi^{(1)}\rangle\), which is the expansion coefficient for the expansion of \(\psi^{(1)}\) in terms of the zeroth-order functions { }. In RSPT, one assumes that the only contribution of \(\psi^{(0)}\) to the full wave function \psioccurs in zeroth-order; this is referred to as assuming intermediate normalization of y. In other words, \(\langle\psi^{(0)}|\psi\rangle = 1\) because \(\langle\psi^{(0)}|\psi^{(0)}\rangle = 1\) and \(\langle\psi^{(0)}|\psi^{(n)}\rangle = 0\) for \(n = 1, 2, 3, \cdots\). So, the coefficients \(\langle\psi_J^{(0)} |\psi^{(1)}\rangle\) appearing in the above equation are all one needs to describe \(\psi^{(1)}\).
3. If the state of interest \(\psi^{(0)}\) is non-degenerate in zeroth-order (i.e., none of the other is equal to E^{(0)}), this equation can be solved for the needed expansion coefficients
\[\langle \psi_J^{(0)}|\psi^{(1)}\rangle=\frac{\langle\psi_J^{(0)} |V|\psi^{(0)}\rangle}{E^{(0)}-E^{(0)}_J}\]
which allow the first-order wave function to be written as
\[\psi^{(1)}=\sum_J\psi_J^{(0)}\frac{\langle\psi_J^{(0)} |V|\psi^{(0)}\rangle}{E^{(0)}-E^{(0)}_J}\]
where the index \(J\) is restricted such that \(\psi_J^{(0)}\) not equal the state \(\psi^{(0)}\) you are interested in.
4. However, if one or more of the zeroth-order energies is equal to \(E^{(0)}\), an additional step needs to be taken before the above expression for \(\psi^{(1)}\) can be used. If one were to try to solve \(\langle \psi_J^{(0)}|\psi^{(1)}\rangle + \langle\psi_J^{(0)} |V|\psi_0\rangle = E^{(0)} \langle \psi_J^{(0)}|\psi^{(1)}\rangle\) without taking this extra step, the \(\langle\psi_J^{(0)} |\psi^{(1)}\rangle\) values for those states with \(= E^{(0)}\) could not be determined because the first and third terms would cancel and the equation would read \(\langle \psi_J^{(0)}|V|\psi^{(0)}\rangle = 0\). The way RSPT deals with this paradox is realize that, within a set of \(N\) degenerate states, any \(N\) orthogonal combinations of these states will also be degenerate. So RSPT assumes that one has already chosen the degenerate sets of zeroth-order states to make \(\langle \psi_J^{(0)}|V| \psi_K^0\rangle = 0\) for \(K \ne J\). This extra step is carried out in practice by forming the matrix representation of \(V\) in the original set of degenerate zeroth-order states and then finding the unitary transformation among these states that diagonalizes this matrix. These transformed states are then what one uses as and \(\psi^{(0)}\) in the RSPT expressions. This means that the paradoxical result \(\langle\psi_J^{(0)}|V|\psi^{(0)}\rangle = 0\) is indeed obeyed by this choice of states, so one does not need to determine the coefficients \(\langle\psi_J^{(0)} |\psi^{(1)}\rangle\) for belonging to the degenerate zeroth-order states (i.e., these coefficients can be assumed to be zero). The bottom line is that the expression
\[\psi^{(1)}=\sum_J\psi_J^{(0)}\frac{\langle\psi_J^{(0)} |V|\psi^{(0)}\rangle}{E^{(0)}-E^{(0)}_J}\]
remains valid, but the summation index \(J\) is now restricted to exclude any members of the zeroth-order states that are degenerate with \(\psi^{(0)}\).
To obtain the expression for the second-order correction to the energy of the state of interest, one returns to
\[H^{(0)} \psi^{(2)} + V \psi^{(1)} = E^{(0)} \psi^{(2)} + E^{(1)} \psi^{(1)} + E^{(2)} \psi^{(0)}\]
Multiplying on the left by the complex conjugate of \(\psi^{(0)}\) and integrating yields
\[\langle\psi^{(0)}|H^{(0)}|\psi^{(2)}\rangle + \langle\psi^{(0)}|V|\psi^{(1)}\rangle = E^{(0)} \langle\psi^{(0)}|\psi^{(2)}\rangle + E^{(1)} \langle\psi^{(0)}|\psi^{(1)}\rangle + E^{(2)} \langle\psi^{(0)}|\psi^{(0)}\rangle.\]
The intermediate normalization condition causes the fourth term to vanish, and the first and third terms cancel one another. Recalling the fact that \(\psi^{(0)}\) is normalized, the above equation reduces to
\[\langle\psi^{(0)}|V|\psi^{(1)}\rangle = E^{(2)}.\]
Substituting the expression obtained earlier for \(\psi^{(1)}\) allows \(E^{(2)}\) to be written as
\[E^{(2)}=\sum_J \frac{|\langle\psi_J^{(0)} |V|\psi^{(0)}\rangle|^2}{E^{(0)}-E^{(0)}_J}\]
where, as before, the sum over \(J\) is limited to states that are not degenerate with \(\psi^{(0)}\) in zeroth-order.
These are the fundamental working equations of Rayleigh-Schrödinger perturbation theory. They instruct us to compute the average value of the perturbation taken over a probability distribution equal to \(\psi^{(0)}{}^*\psi^{(0)}\) to obtain the first-order correction to the energy \(E^{(1)}\). They also tell us how to compute the first-order correction to the wave function and the second-order energy in terms of integrals coupling \(\psi^{(0)}\) to other zeroth-order states and denominators involving energy differences .
An analogous approach is used to solve the second- and higher-order equations. For example, the equation for the nth order energy and wave functions reads:
\[H^{(0)} \psi^{(n)} + V \psi^{(n-1)} = E^{(0)} \psi^{(n)} + E^{(1)} \psi^{(n-1)} + E^{(2)} \psi^{(n-2)} + E^{(3)} \psi^{(n-3)} + … + E^{(n)} \psi^{(0)}\]
The nth order energy is obtained by multiplying this equation on the left by \(\psi^{(0)}{}^*\) and integrating over the relevant coordinates (and using the fact that \(\psi^{(0)}\) is normalized and the intermediate normalization condition \(\langle\psi^{(0)}|\psi_m\rangle = 0\) for all \(m > 0\)):
\[\langle\psi^{(0)}|V|\psi^{(n-1)}\rangle = E^{(n)}.\]
This allows one to recursively solve for higher and higher energy corrections once the various lower-order wave functions \(\psi^{(n-1)}\) are obtained. To obtain the expansion coefficients for the \(\psi^{(n)}\) expanded in terms of the zeroth-order states { }, one multiplies the above \(n^{th}\) order equation on the left by (one of the zeroth-order states not equal to the state \(\psi^{(0)}\) of interest) and obtains
\[\langle\psi_J^{(0)} |\psi^{(n)}\rangle + \langle\psi_J^{(0)} |V| \psi^{(n-1)}\rangle = E^{(0)} \langle\psi_J^{(0)} |\psi^{(n)}\rangle + E^{(1)} \langle \psi_J^{(0)}|\psi^{(n-1)}\rangle + E^{(2)} \langle \psi_J^{(0)}|\psi^{(n-2)}\rangle + E^{(3)} \langle\psi_J^{(0)} |\psi^{(n-3)}\rangle + … + E^{(n)} \langle\psi_J^{(0)} |\psi^{(0)}\rangle.\]
The last term on the right-hand side vanishes because and \(\psi^{(0)}\) are orthogonal. The terms containing the nth order expansion coefficients \(\langle |\psi^{(n)}\rangle\) can be brought to the left-hand side to produce the following equation for these unknowns:
\[\langle\psi_J^{(0)} |\psi^{(n)}\rangle - E^{(0)} \langle\psi_J^{(0)} |\psi^{(n)}\rangle = - \langle \psi_J^{(0)}|V| \psi^{(n-1)}\rangle + E^{(1)} \langle\psi_J^{(0)} |\psi^{(n-1)}\rangle + E^{(2)} \langle\psi_J^{(0)}|\psi^{(n-2)}\rangle + E^{(3)} \langle \psi_J^{(0)}|\psi^{(n-3)}\rangle + … + E^{(n)} \langle \psi_J^{(0)}|\psi^{(0)}\rangle.\]
As long as the zeroth-order energy is not degenerate with \(E^{(0)}\) (or, that the zeroth-order states have been chosen as discussed earlier to cause there to no contribution to \(\psi^{(n)}\) from such degenerate states), the above equation can be solved for the expansion coefficients \(\langle \psi_J^{(0)}|\psi^{(n)}\rangle\), which then define \(\psi^{(n)}\).
The RSPT equations can be solved recursively to obtain even high-order energy and wave function corrections:
- \(\psi^{(0)}\) and \(E^{(0)}\) and \(V\) are used to determine \(E^{(1)}\) and \(\psi^{(1)}\) as outlined above,
- \(E^{(2)}\) is determined from \(\langle\psi_0|V|\psi_{n-1}\rangle = E^{(n)}\) with \(n = 2\), and the expansion coefficients of \(\psi^{(2)}\) {\(\langle |\psi^{(2)}\rangle\)} are determined from the above equation with \(n = 2\),
- \(E^{(3)}\) (and higher \(E^{(n)}\)) are then determined from \(\langle\psi_0|V|\psi_{n-1}\rangle = E^{(n)}\) and the expansion coefficients of \(\psi^{(2)}\) {\(\langle |\psi^{(2)}\rangle\)} are determined from the above equation with \(n = 2\).
- This process can then be continued to higher and higher order.
Although modern quantum mechanics uses high-order perturbation theory in some cases, much of what the student needs to know is contained in the first- and second- order results to which I will therefore restrict our further attention. I recommend that students have in memory (their own brain, not a computer) the equations for \(E^{(1)}\), \(E^{(2)}\), and \(\psi^{(1)}_0\) so they can make use of them even in qualitative applications of perturbation theory as we will discuss later in this Chapter. But, first, let’s consider an example problem that illustrates how perturbation theory is used in a more quantitative manner.
Example Problem
As we discussed earlier, an electron moving in a quasi-linear conjugated bond framework can be modeled as a particle in a box. An externally applied electric field of strength \(\varepsilon\) interacts with the electron in a fashion that can described by adding the perturbation \(V = e\varepsilon\Big(x-\dfrac{L}{2}\Big)\) to the zeroth-order Hamiltonian. Here, \(x\) is the position of the electron in the box, \(e\) is the electron's charge, and \(L\) is the length of the box. The perturbation potential varies in a linear fashion across the box, so it acts to pull the electron to one side of the box.
First, we will compute the first-order correction to the energy of the \(n=1\) state and the first-order wave function for the \(n=1\) state. In the wave function calculation, we will only compute the contribution to \(\psi\) made by \(\psi^{(0)}_2\) (this is just an approximation to keep things simple in this example). Let me now do all the steps needed to solve this part of the problem. Try to make sure you can do the algebra, but also make sure you understand how we are using the first-order perturbation equations.
The zeroth-order wave functions and energies are given by
\[\psi^{(0)}_n= \sqrt{\frac{2}{L}}\sin\left(\frac{n\pi x}{L}\right) , \]
and
\[E^{(0)}_n= \frac{\hbar^2\pi^2 n^2}{2mL^2},\]
and the perturbation is
\[V = e\varepsilon\left(x-\frac{L}{2}\right).\]
The first-order correction to the energy for the state having \(n = 1\) and denote \(\psi^{(0)}\) is
\[E^{(1)} =\langle\psi^{(0)}|V|\psi^{(0)}\rangle =\left\langle\psi^{(0)}\left|e\varepsilon\left(x-\frac{L}{2}\right)\right|\psi^{(0)}\right\rangle\]
\[=\frac{2}{L}\int_0^L \sin^2\left(\frac{\pi x}{L}\right)e\varepsilon\left(x-\frac{L}{2}\right)dx\]
\[=\frac{2e\varepsilon}{L}\int_0^L\sin^2\left(\frac{\pi x}{L}\right)xdx -\frac{2e\varepsilon}{L}\frac{L}{2}\int_0^L\sin^2\left(\frac{\pi x}{L}\right)dx\]
The first integral can be evaluated using the following identity with \(a = \dfrac{\pi}{L}\):
\[\int_0^L\sin^2(ax)dx=\frac{x^2}{4}-\frac{x\sin(2ax)}{4a}-\frac{x\cos(2ax)}{8a^2}\Bigg|^L_0=\frac{L^2}{4}\]
The second integral can be evaluated using the following identity with \(\theta =\frac{\pi x}{L}\)
and \(d\theta = \frac{\pi}{L}dx\) :
\[\int_0^L\sin^2\left(\frac{\pi x}{L}\right)dx= \frac{L}{\pi}\int_0^\pi\sin^2\theta d\theta=-\frac{1}{4}\sin(2\theta) + \frac{\theta}{2}\Bigg|^\pi_0=\frac{\pi}{2} \].
Making all of these appropriate substitutions we obtain:
\[E^{(1)} =\frac{2e\varepsilon}{L}\left(\frac{L^2}{4}-\frac{L}{2}\frac{L}{\pi}\frac{\pi}{2}\right) = 0.\]
This result, that the first-order correction to the energy vanishes, could have been foreseen. In the expression for \(E^{(1)} = \langle\psi^{(0)}|V|\psi^{(0)}\rangle\), the product \(\psi^{(0)}{}^*\psi^{(0)}\) is an even function under reflection of \(x\) through the midpoint \(x = \dfrac{L}{2}\); in fact, this is true for all of the particle-in-a-box wave functions. On the other hand, the perturbation \(V = e\varepsilon\Big(x-\dfrac{L}{2}\Big)\) is an odd function under reflection through \(x = \dfrac{L}{2}\). Thus, the integral \(\langle\psi^{(0)}|V|\psi^{(0)}\rangle\) must vanish as its integrand is an odd function. This simple example illustrates how one can use symmetry to tell ahead of time whether the integrals \(\langle\psi^{(0)}|V|\psi^{(0)}\rangle\) and \(\langle\psi_J^{(0)}|V|\psi^{(0)}\rangle\) contributing to the first-order and higher-order energies and wave functions will vanish.
The contribution to the first-order wave function made by the \(n = 2\) state is given by
\[\psi^{(1)}=\frac{\langle\psi_J^{(0)} |V|\psi^{(0)}_2\rangle\psi^{(0)}_2}{E^{(0)}-E^{(0)}_2}\]
\[=\frac{\dfrac{2}{L}\left\langle\sin\left(\dfrac{\pi x}{L}\right)\left|e\varepsilon\left(x-\frac{L}{2}\right)\right|\sin\left(\dfrac{2 \pi x}{L}\right)\right\rangle}{\dfrac{\hbar^2\pi^2}{2mL^2}-\dfrac{\hbar^2\pi^2 2^2}{2mL^2}}\]
The two integrals in the numerator involve
\[\int_0^L x\sin\left(\dfrac{2 \pi x}{L}\right)\sin\left(\dfrac{\pi x}{L}\right)dx\]
and
\[\int_0^L \sin\left(\dfrac{2 \pi x}{L}\right)\sin\left(\dfrac{\pi x}{L}\right)dx\]
Using the integral identities
\[\int x\cos(ax)dx= \frac{1}{a^2}\cos(ax) + \frac{x}{a} \sin(ax)\]
and
\[\int\cos(ax)dx = \frac{1}{a}\sin(ax),\]
we obtain the following:
\[\int_0^L \sin\left(\dfrac{2 \pi x}{L}\right)\sin\left(\dfrac{\pi x}{L}\right)dx=\frac{1}{2}\left[\int_0^L \cos\left(\dfrac{\pi x}{L}\right)dx - \int_0^L \cos\left(\dfrac{3\pi x}{L}\right)dx\right]\]
\[=\frac{1}{2}\left[ \frac{L}{\pi}\sin\left(\dfrac{\pi x}{L}\right)\Bigg|^L - \frac{L}{3\pi}\sin\left(\dfrac{3\pi x}{L}\right)\Bigg|^L\right] = 0\]
and
\[\int_0^L x\sin\left(\dfrac{2 \pi x}{L}\right)\sin\left(\dfrac{\pi x}{L}\right)dx=
\frac{1}{2}\left[\int_0^L x\cos\left(\dfrac{\pi x}{L}\right)dx - \int_0^L x\cos\left(\dfrac{3\pi x}{L}\right)dx\right]\]
\[=\frac{1}{2}\left[ \left(\frac{L^2}{\pi^2}\cos\left(\dfrac{\pi x}{L}\right)+ \frac{Lx}{\pi}\sin\left(\dfrac{\pi x}{L}\right) \right)\Bigg|^L
- \left(\frac{L^2}{9\pi^2}\cos\left(\dfrac{3\pi x}{L}\right)+ \frac{Lx}{3\pi}\sin\left(\dfrac{3\pi x}{L}\right)\right) \Bigg|^L\right] \]
\[=\frac{-2L^2}{2\pi^2} -\frac{-2L^2}{18\pi^2} = \frac{L^2}{9\pi^2} -\frac{L^2}{\pi^2} = - \frac{-8L^2}{9\pi^2}.\]
Making all of these appropriate substitutions we obtain:
\[\psi^{(1)}=\frac{32mL^3e\varepsilon}{27\hbar^2\pi^4}\sqrt{\frac{2}{L}}\sin\left(\frac{2\pi x}{L}\right)\]
for the first-order wave function (actually, only the \(n = 2\) contribution). So, the wave function through first order (i.e., the sum of the zeorth- and first-order pieces) is
\[\psi^{(0)}+\psi^{(1)}=\sqrt{\frac{2}{L}}\sin\left(\frac{\pi x}{L}\right) + \frac{32mL^3e\varepsilon}{27\hbar^2\pi^4}\sqrt{\frac{2}{L}}\sin\left(\frac{2\pi x}{L}\right)\]
In Figure 4.1 we show the \(n = 1\) and \(n = 2\) zeroth-order functions as well as the superposition function formed when the zeroth-order \(n = 1\) and first-order \(n = 1\) functions combine.
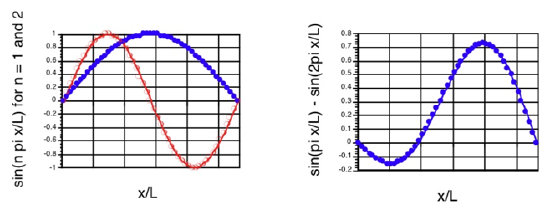
Clearly, the external electric field acts to polarize the \(n = 1\) wave function in a manner that moves its probability density toward the \(x > \dfrac{L}{2}\) side of the box. The degree of polarization will depend on the strength of the applied electric field.
For such a polarized superposition wave function, there should be a net dipole moment induced in the system. We can evaluate this dipole moment by computing the expectation value of the dipole moment operator:
\[\mu_{induced}= - e \int\psi^*\left(x-\frac{L}{2}\right)\psi dx\]
with being the sum of our zeroth- and first-order wave functions. In computing this integral, we neglect the term proportional to \(E^{(2)}\) because we are interested in only the term linear in \(\varepsilon\) because this is what gives the dipole moment. Again, allow me to do the algebra and see if you can follow.
\[\mu_{induced}= - e \int\psi^*\left(x-\frac{L}{2}\right)\psi dx\]
where,
\[\psi=\psi^{(0)}+\psi^{(1)}\]
\[\mu_{induced}= - e \int_0^L(\psi^{(0)}+\psi^{(1)})^*\left(x-\frac{L}{2}\right)(\psi^{(0)}+\psi^{(1)}) dx\]
\[= - e \int_0^L \psi^{(0)}{}^*\left(x-\frac{L}{2}\right)\psi^{(0)} dx - e \int_0^L \psi^{(1)}{}^*\left(x-\frac{L}{2}\right)\psi^{(0)} dx\]
\[= - e \int_0^L \psi^{(0)}{}^*\left(x-\frac{L}{2}\right)\psi^{(1)} dx - e \int_0^L \psi^{(1)}{}^*\left(x-\frac{L}{2}\right)\psi^{(1)} dx\]
The first integral is zero (we discussed this earlier when we used symmetry to explain why this vanishes). The fourth integral is neglected since it is proportional to \(E^{(2)}\) and we are interested in obtaining an expression for how the dipole varies linearly with \(\varepsilon\). The second and third integrals are identical and can be combined to give:
\[\mu_{induced}= - 2 e \int_0^L \psi^{(0)}{}^*\left(x-\frac{L}{2}\right)\psi^{(1)} dx\]
Substituting our earlier expressions for
\[\psi^{(0)}=\dfrac{2}{L}\sin\left(\dfrac{\pi x}{L}\right)\]
and
\[\psi^{(1)}=\frac{32mL^3e\varepsilon}{27\hbar^2\pi^4}\sqrt{\frac{2}{L}}\sin\left(\frac{2\pi x}{L}\right)\]
we obtain:
\[\mu_{induced} = -2e\frac{32mL^3e\varepsilon}{27\hbar^2\pi^4} \frac{2}{L} \int_0^L \sin\left(\dfrac{\pi x}{L}\right) \left(x-\frac{L}{2}\right) \sin\left(\dfrac{2\pi x}{L}\right) dx \]
These integrals are familiar from what we did to compute ; doing them we finally obtain:
\[\mu_{induced}= -2e \frac{32mL^3e\varepsilon}{27\hbar^2\pi^4} \left(\frac{2}{L}\right) \left(\frac{-8L^2}{9\pi^2}\right) =\frac{mL^4 e^2\varepsilon}{\hbar^2\pi^6} \frac{2^{10}}{3^5}\]
Now. Let’s compute the polarizability, \(\alpha\), of the electron in the \(n=1\) state of the box, and try to understand physically why a should depend as it does upon the length of the box \(L\). To compute the polarizability, we need to know that
\[\alpha = \left(\frac{\partial\mu}{\partial\varepsilon}\right)_{\varepsilon=0}.\]
Using our induced moment result above, we then find
\[\alpha =\left(\frac{\partial\mu}{\partial\varepsilon}\right)_{\varepsilon=0} = \frac{mL^4 e^2}{\hbar^2\pi^6} \frac{2^{10}}{3^5}\]
Notice that this finding suggests that the larger the box (i.e., the length of the conjugated molecule), the more polarizable the electron density. This result also suggests that the polarizability of conjugated polyenes should vary non-linearly with the length of the conjugated chain.
Other Examples
Let’s consider a few more examples of how perturbation theory is used in chemistry, either quantitatively (i.e., to actually compute changes in energies and wave functions) or qualitatively (i.e., to interpret or anticipate how changes might alter energies or other properties).
The Stark effect
When a molecule is exposed to an electric field \(\textbf{E}\), its electrons and nuclei experience a perturbation
\[V= \textbf{E} \cdot ( e\sum_n Z_n \textbf{R}_n - e \sum_i \textbf{r}_i )\]
where \(Z_n\) is the charge of the \(n^{th}\) nucleus whose position is \(R_n\), \(r_i\) is the position of the \(i^{th}\) electron, and \(e\) is the unit of charge. The effect of this perturbation on the energies is termed the Stark effect. The first-order change to the energy of this molecule is evaluated by calculating
\[E^{(1)}=\langle\psi^*|V|\psi\rangle=\textbf{E}\cdot \langle\psi|e\sum_n Z_n \textbf{R}_n-e\sum_i \textbf{r}_i|\psi\]
where \(\psi\) is the unperturbed wave function of the molecule (i.e., the wave function in the absence of the electric field). The quantity inside the integral is the electric dipole operator, so this integral is the dipole moment of the molecule in the absence of the field. For species that possess no dipole moment (e.g., non-degenerate states of atoms and centro-symmetric molecules), this first-order energy vanishes. It vanishes in the two specific cases mentioned because \(\psi\) is either even or odd under the inversion symmetry, but the product \(\psi^* \psi\) is even, and the dipole operator is odd, so the integrand is odd and thus the integral vanishes.
If one is dealing with a degenerate state of a centro-symmetric system, things are different. For example, the \(2s\) and \(2p\) states of the hydrogen atom are degenerate, so, to apply perturbation theory one has to choose specific combinations that diagonalize the perturbation. This means one needs to first form the 2x2 matrix
\[\left(\begin{array}{cc}
\langle 2s |V| 2s \rangle & \langle 2s |V| 2p_z \rangle \\
\langle 2p_z |V| 2s \rangle & \langle 2p_z |V| 2p_z \rangle
\end{array}\right)
\]
where \(z\) is taken to be the direction of the electric field. The diagonal elements of this matrix vanish due to parity symmetry, so the two eigenvalues are equal to
\[E^{(1)}_{\pm}=\pm2\langle 2s|V| 2p_z \rangle.\]
These are the two first-order (because they are linear in \(V\) and thus linear in ) energies.
So, in such degenerate cases, one can obtain linear Stark effects. The two corrected zeroth-order wave functions corresponding to these two shifted energies are
\[\psi^{(0)}_{\pm}=\frac{1}{\sqrt{2}}[2s\mp 2p_z]\]
and correspond to orbitals polarized into or away from the electric field.
The Stark effect example offers a good chance to explain a fundamental problem with applying perturbation theory. One of the basic assumptions of perturbation theory is that the unperturbed and perturbed Hamiltonians are both bounded from below (i.e., have a discrete lowest eigenvalues) and allow each eigenvalue of the unperturbed Hamiltonian to be connected to a unique eigenvalue of the perturbed Hamiltonian. Considering the example just discussed, we can see that these assumptions are not met for the Stark perturbation.
Consider the potential that an electron experiences within an atom or molecule close to a nucleus of charge \(Z\). It is of the form (in atomic units where the energy is given in Hartrees (1 H = 27.21 eV) and distances in Bohr units (1 Bohr = 0.529 Å))
\[V(r,\theta,\phi)=-\frac{Z}{r}-e\textbf{E}r\cos\theta\]
where the first term is the Coulomb potential acting to attract the electron to the nucleus and the second is the electron-field potential assuming the field is directed along the \(z\)-direction. In Figure 4.2 a we show this potential for a given value of the angle \(\theta\).
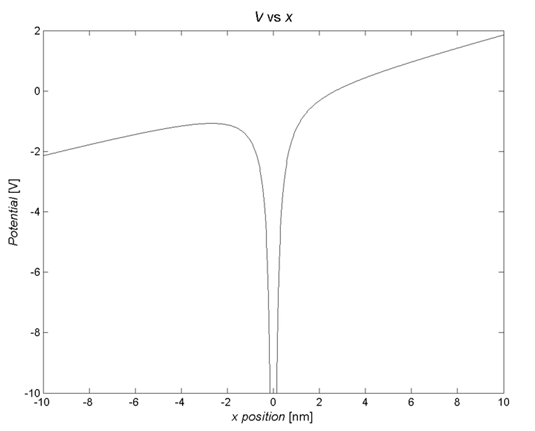
Along directions for which \(\cos{\theta}\) is negative (to the right in Figure 4.2 a), this potential becomes large and positive as the distance \(r\) of the electron from the nucleus increases; for bound states such as the \(2s\) and \(2p\) states discussed earlier, such regions are classically forbidden and the wave function exponentially decays in this direction. However, in directions along which \(\cos{\theta}\) is positive, the potential is negative and strongly attractive for small-r (i.e., near the nucleus), then passes through a maximum (e.g., near \(x = -2\) in Figure 4.2 a) at
\[r_{\rm max}=\sqrt{\frac{Z}{e\textbf{E}\cos\theta}}\]
where
\[V(r_{\rm max})=-2\sqrt{e\textbf{E}\cos\theta}\]
(ca. – 1 eV in Figure 4.2 a) and then decreases monotonically as r increases. In fact, this potential approaches \(-\infty\) as \(r\) approaches \(\infty\) as we see in the left portion of Figure 4.2 a.
The bottom line is that the total potential with the electric field present violates the assumptions on which perturbation theory is based. However, it turns out that perturbation theory can be used in such cases under certain conditions. For example as applied to the Stark effect for the degenerate \(2s\) and \(2p\) levels of a hydrogenic atom (i.e., a one-electron system with nuclear charge \(Z\)), if the energy of the \(2s\) and \(2p\) states lies far below the maximum in the potential \(V(r_{\rm max})\), perturbation theory can be used. We know the energies of hydrogenic ions vary with \(Z\) and with the principal quantum number \(n\) as
\[E^{(n)}(Z)=\frac{-13.6 {\rm eV}}{n^2Z^2}=\frac{-1}{2n^2Z^2}{\rm au}\]
So, as long as
\[\frac{-1}{2n^2Z^2} \ll -2\sqrt{e\textbf{E}\cos\theta}\]
the zeroth-order energy of the state will like below the barrier on the potential surface. Because the wave function can penetrate this barrier, this state will no longer be a true bound state; it will be a metastable resonance state (recall, we studied such states in Chapter 1 where we learned about tunneling). However, if the zeroth-order energy lies far below the barrier, the extent of tunneling through the barrier will be small, so the state will have a long lifetime. In such cases, we can use perturbation theory to describe the effects of the applied electric field on the energies and wave functions of such metastable states, but we must realize that we are only allowed to do so in the limit of weak fields and for states that lie significantly below the barrier. In this case, perturbation theory describes the changes in the energy and wave function in regions of space where the zeroth-order wave function is bound, but does not describe at all the asymptotic part of the wave function where the electron is unbound.
Another example of Stark effects in degenerate cases arises when considering how polar diatomic molecules’ rotational energies are altered by an electric field. The zeroth-order wave functions appropriate to such cases are given by
\[\psi=Y_{J,M}(\theta,\phi)\chi_\nu(R)\psi_e(r|R)\]
where the spherical harmonic \(Y_{J,M}(\theta,\phi)\) is the rotational wave function, \(\chi_\nu(R)\) is the vibrational function for level \(\nu\), and \(\psi_e(r|R)\) is the electronic wave function. The diagonal elements of the electric-dipole operator
\[\langle Y_{J,M}(\theta,\phi)\chi_\nu(R)\psi_e(r|R)|V|Y_{J,M}(\theta,\phi)\chi_\nu(R)\psi_e(r|R) \rangle\]
vanish because the vibrationally averaged dipole moment, which arises as
\[\langle\mu\rangle = \langle \chi_\nu(R)\psi_e(r|R)| e\sum_n Z_n \textbf{R}_n-e\sum_i \textbf{r}_i |\chi_\nu(R)\psi_e(r|R) \rangle\]
is a vector quantity whose component along the electric field is \(\langle \mu\rangle \cos(\theta)\) (again taking the field to lie along the \(z\)-direction). Thinking of \(\cos(\theta)\) as \(x\), so \(\sin(\theta) d\theta\) is \(dx\), the integrals
\[\langle Y_{J,M}(\theta,\phi)|\cos\theta|Y_{J,M}(\theta,\phi)\rangle=\int Y_{J,M}^*(\theta,\phi) \cos\theta Y_{J,M}(\theta,\phi) \sin\theta d\theta d\phi=\int Y_{J,M}^*(\theta,\phi) x Y_{J,M}(\theta,\phi) dxd\phi=0\]
because \(|Y_{J,M}|^2\) is an even function of \(x\) (i.e. ,of \(\cos(\theta)\)). Because the angular dependence of the perturbation (i.e., \(\cos \theta\)) has no \(\phi\)-dependence, matrix elements of the form
\[ \int Y_{J,M}^*(\theta,\phi) \cos\theta Y_{J,M}(\theta,\phi)\sin\theta d\theta d\phi=0\]
also vanish. This means that if one were to form the \((2J+1) \times (2J+1)\) matrix representation of \(V\) for the \(2J+1\) degenerate states \(Y_{J,M}\) belonging to a given \(J\), all of its elements would be zero. Thus the rotational energies of polar diatomic (or rigid linear polyatomic) molecules have no first-order Stark splittings.
There will, however, be second-order Stark splittings, in which case we need to examine the terms that arise in the formula
\[E^{(2)}=\sum_J\frac{|\langle\psi^{(0)}|V|psi^{(0)}\rangle|^2}{E^{(0)}-E^{(0)}_J}\]
For a zeroth-order state \(Y_{J,M}\), only certain other zeroth-order states will have non-vanishing coupling matrix elements . These non-zero integrals are governed by , which can be shown to be
\[\langle Y_{J,M}|\cos\theta|Y_{J',M'}\rangle=\sqrt{\frac{(J+1)^2-M^2}{(2J+1)(2J+3)}}\delta_{M,M'}\text{ for }J'=J+1; \sqrt{\frac{J^2-M^2}{(2J-1)(2J+1)}}\delta_{M,M'}\text{ for }J'=J-1;\]
of course, if \(J = 0\), the term \(J’ = J-1\) does not occur. The limitation that \(M\) must equal \(M’\) arises, as above, because the perturbation contains no terms dependent on the variable \(\phi\). The limitation that \(J’ = J±1\) comes from a combination of three conditions
- angular momentum coupling, which you learned about in Chapter 2, tells us that \(\cos\theta\), which happens to be proportional to \(Y_{1,0}(\theta,\phi)\), can couple to \(Y_{J,M}\) to generate terms having \(J+1\), \(J\), or \(J-1\) for their \(J^2\) quantum number but only \(M\) for their \(J_z\) quantum number,
- the \(J+1\), \(J\), and \(J-1\) factors arising from the product \(\cos \theta Y_{J,M}\) must match \(Y_{J,M'}\) for the integral not to vanish because \(\langle Y_{J,M}|Y_{J',M'}\rangle = \delta_{J,J’} \delta_{M,M’}\),
- finally, the \(J = J’\) terms will vanish because of the inversion symmetry (\(\cos\theta\) is odd under inversion but \(|Y_{J,M}|^2\) is even).
Using the fact that the perturbation is \(\textbf{E}\langle \mu\rangle \cos(theta)\), these two non-zero matrix elements can be used to express the second-order energy for the \(J,M\) level as
\[E=\textbf{E}^2\langle\mu\rangle^2\left[\dfrac{\dfrac{(J+1)^2-M^2}{(2J+1)(2J+3)}}{-2B(J+1)} +
\dfrac{\dfrac{J^2-M^2}{(2J-1)(2J+1)}}{2BJ}\right]\]
where \(h\) is Planck’s constant and \(B\) is the rotational constant for the molecule
\[B=\frac{h}{8\pi^2\mu r_e^2}\]
for a diatomic molecule of reduced mass \(\mu\) and equilibrium bond length \(r_e\).
Before moving on to another example, it is useful to point out some common threads that occur in many applications of perturbation theory and that will also be common to variational calculations that we discuss later in this Chapter. Once one has identified the specific zeroth-order state \(\psi^{(0)}\) of interest, one proceeds as follows:
- The first-order energy \(E^{(1)} = \langle \psi^{(0)}|V| \psi^{(0)}\rangle\) is evaluated. In doing so, one should first make use of any symmetry (point group symmetry is treated later in this Chapter) such as inversion, angular momentum, spin, etc., to determine whether this expectation value will vanish by symmetry, in which case, we don’t bother to consider this matrix element any more. We used this earlier when considering \(\langle 2s|\cos\theta|2s\rangle\), \(\langle2p_\sigma|\cos\theta|2p_\sigma\rangle\), and \(\langle Y_{J,M}|\cos\theta|Y_{J,M}\rangle\) to conclude that certain first-order energies are zero.
- If \(E^{(1)}\) vanishes (so the lowest-order effect is in second order) or if we want to examine higher-order corrections, we consider evaluating \(E^{(2)}\). Before doing so explicitly, we think about whether symmetry will limit the matrix elements \(\langle\psi^{(0)}|V \psi^{(0)}n\rangle\) entering into the expression for \(E^{(2)}\). For example, in the case just studied, we saw that only other zeroth-order states having \(J’ = J+1\) or \(J‘ = J-1\) gave non-vanishing matrix elements. In addition, because \(E^{(2)}\) contains energy denominators (\(E^{(0)}-E^{(0)}_n\)), we may choose to limit our calculation to those other zeroth-order states whose energies are close to our state of interest; this assumes that such states will contribute a dominant amount to the sum
.
You will encounter many times when reading literature articles in which perturbation theory is employed situations in which researchers have focused attention on zeroth-order states that are close in energy to the state of interest and that have the correct symmetry to couple strongly (i.e., have substantial \(\langle \psi^{(0)}|V \psi^{(0)}_n\rangle\)) to that state.
Electron-electron Coulomb repulsion
In one of the most elementary pictures of atomic electronic structure, one uses nuclear charge screening concepts to partially account for electron-electron interactions. For example, in 1s22s1 Li, one might posit a zeroth-order wave function consisting of a product
\[\psi=\phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3)\]
in which two electrons occupy a \(1s\) orbital and one electron occupies a \(2s\) orbital. To find a reasonable form for the radial parts of these two orbitals, one could express each of them as a linear combination of (i) one orbital having hydrogenic \(1s\) form with a nuclear charge of 3 and (ii) a second orbital of \(2s\) form but with a nuclear charge of 1 (to account for the screening of the \(Z = 3\) nucleus by the two inner-shell \(1s\) electrons)
\[\phi_i(r)=C_i\chi_{1s,Z=1}(r)+D_i\chi_{2s,Z=3}(r)\]
where the index i labels the \(1s\) and \(2s\) orbitals to be determined. Next, one could determine the \(C_i\) and \(D_i\) expansion coefficients by requiring the fi to be approximate eigenfunctions of the Hamiltonian
\[h=\frac{1}{2}\nabla^2-\frac{3}{r}\]
that would be appropriate for an electron attracted to the Li nucleus but not experiencing any repulsions with other electrons. This would result in the following equation for the expansion coefficients:
\[
\left(\begin{array}{cc}
\langle \chi_{1s,Z=1}(r) | -\frac{1}{2}\nabla^2-\frac{3}{r}|\chi_{1s,Z=1}(r) \rangle
& \langle \chi_{1s,Z=1}(r) | -\frac{1}{2}\nabla^2-\frac{3}{r}| \chi_{2s,Z=3}(r) \rangle \\
\langle \chi_{1s,Z=1}(r) | -\frac{1}{2}\nabla^2-\frac{3}{r}| \chi_{2s,Z=3}(r) \rangle
& \langle \chi_{2s,Z=3}(r) | -\frac{1}{2}\nabla^2-\frac{3}{r}| \chi_{2s,Z=3}(r) \rangle
\end{array}\right)
\left(\begin{array}{cc}C \\ D\end{array}\right)\\
=
\left(\begin{array}{cc}
\langle \chi_{1s,Z=1}(r) | \chi_{1s,Z=1}(r) \rangle & \langle \chi_{1s,Z=1}(r) | \chi_{2s,Z=3}(r) \rangle \\
\langle \chi_{1s,Z=1}(r) | \chi_{2s,Z=3}(r) \rangle & \langle \chi_{2s,Z=3}(r) | \chi_{2s,Z=3}(r) \rangle
\end{array}\right)
\left(\begin{array}{cc}C \\ D\end{array}\right).
\]
This 2x2 matrix eigenvalue problem can be solved for the \(C_i\) and \(D_i\) coefficients and for the energies \(E_i\) of the \(1s\) and \(2s\) orbitals. The lower-energy solution will have \(|C| > |D|\), and will be this model’s description of the \(1s\) orbital. The higher-energy solution will have \(|D| > |C|\) and is the approximation to the \(2s\) orbital.
Using these \(1s\) and \(2s\) orbitals and the 3-electron wave function they form
\[\psi=\phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3)\]
as a zeroth-order approximation, how do we then proceed to apply perturbation theory? The full three-electron Hamiltonian
\[H=\sum_{i=1}^3\left[\frac{1}{2}\nabla_i^2-\frac{3}{r_i}\right]+\sum_{i<j=1}^3\frac{1}{r_{i,j}}\]
can be decomposed into a zeroth-order part
\[H^{(0)}=\sum_{i=1}^3\left[\frac{1}{2}\nabla_i^2-\frac{3}{r_i}\right]\]
and a perturbation
\[V=\sum_{i<j=1}^3\frac{1}{r_{i,j}}\]
The zeroth-order energy of the wave function
\[\psi=\phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3)\]
is
\[E^{(0)}=2E_{1s}+E_{2s}\]
where each of the \(E_{ns}\) are the energies obtained by solving the 2x2 matrix eigenvalue equation shown earlier. The first-order energy of this state can be written as
\[E^{(1)}=\langle \phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3)|V|\phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3) \rangle
J_{1s,1s}+2J_{1s,2s}\]
with the Coulomb interaction integrals being defined as
\[J_{a,b}=\int \phi_a^*(r)\phi_a(r)\frac{1}{|r-r'|}\phi_b^*(r)\phi_b(r)drdr'\]
To carry out the 3-electron integral appearing in \(E^{(1)}\), one proceeds as follows. For the integral
\[\int[\phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3)]^*\frac{1}{r_{1,2}}\phi_{1s}(r_1)\alpha(1)\phi_{1s}(r_2)\beta(2)\phi_{2s}(r_3)\alpha(3)dr_1dr_2dr_3\]
one integrates over the 3 spin variables using \(\langle a| a\rangle=1\), \(\langle a| b\rangle=0\) and \(\langle b| b\rangle=1\)) and then integrates over the coordinate of the third electron using \(\langle \phi_{2s}|\phi_{2s}\rangle=1\) to obtain
\[\int [\phi_{1s}(r_1)\phi_{1s}(r_2)]^*\frac{1}{r_{1,2}} \phi_{1s}(r_1)\phi_{1s}(r_2)dr_1dr_2dr_3\]
which is \(J_{1s,1s}\). The two \(J_{1s,2s}\) integrals arise when carrying out similar integration for the terms arising from (\(1/r_{1,3}\) ) and (\(1/r_{2,3}\)).
So, through first order, the energy of the Li atom at this level of treatment is given by
\[E^{(0)}+E^{(1)}=2E_{1s}+E_{2s}+J_{1s,1s}+2J_{1s,2s}.\]
The factor \(2E_{1s}+E_{2s}\) contains the contributions from the kinetic energy and electron-nuclear Coulomb potential. The \(J_{1s,1s}+2J_{1s,2s}\) terms describe the Coulombic repulsions among the three electrons. Each of the Coulomb integrals \(J_{i,j}\) can be interpreted as being equal to the Coulombic interaction between electrons (one at location \(\textbf{r}\); the other at \(\textbf{r}’\)) averaged over the positions of these two electrons with their spatial probability distributions being given by \(|\phi_i(r)|^2\) and \(|\phi_j(r’)|^2\), respectively.
Although the example just considered is rather primitive, it introduces a point of view that characterizes one of the most commonly employed models for treating atomic and molecular electronic structure- the Hartree-Fock (HF) mean-field model, which we will discuss more in Chapter 6. In the HF model, one uses as a zeroth-order Hamiltonian
\[H^{(0)}=\sum_{i=1}^3 \left[\frac{1}{2}\nabla_i^2-\frac{3}{r_i}+V_{\rm HF}(r_i)\right]\]
consisting of a sum of one-electron terms containing the kinetic energy, the Coulomb attraction to the nucleus (I use the Li atom as an example here), and a potential \(V_{\rm HF}(\textbf{r}_i)\). This potential, which is written in terms of Coulomb integrals similar to those we discussed earlier as well as so-called exchange integrals that we will discuss in Chapter 6, is designed to approximate the interaction of an electron at location \(\textbf{r}_i\) with the other electrons in the atom or molecule. Because \(H^{(0)}\) is one-electron additive, its eigenfunctions consist of products of eigenfunctions of the operator
\[h^{(0)}=\frac{1}{2}\nabla^2-\frac{3}{r}+V_{\rm HF}(r)\]
\(V_{\rm HF}(\textbf{r}_i)\) offers an approximation to the true \(1/r_{i,j}\) Coulomb interactions expressed in terms of a “smeared-out” electron distribution interacting with the electron at ri. Perturbation theory is then used to treat the effect of the perturbation
\[V=\sum_{i<j=1}^N\frac{1}{r_{i,j}}-\sum_{i=1}^N V_{\rm HF}(r_i)\]
on the zeroth-order states. We say that the perturbation, often called the fluctuation potential, corrects for the difference between the instantaneous Coulomb interactions among the \(N\) electrons and the mean-field (average) interactions.
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)