
17.3: Enzymatic reactions with free radical intermediates
- Page ID
- 1022

The radical chain reactions that we have seen so far in this chapter have all been uncatalyzed. Many enzymatic reactions also occur with single-electron mechanistic steps and free radical intermediates. Enzyme-catalyzed radical processes, however, are of course highly specific chemical events.
17.3A: Hydroxylation of alkanes
Radical mechanisms are most relevant in transformations involving functional groups that are relatively unreactive, in particular isolated alkyl carbons. None of the two-electron mechanisms that we have studied so far, for example, could be invoked for the hydroxylation of an alkane, such as this reaction in the biosynthesis of the steroid hormone cortisol.
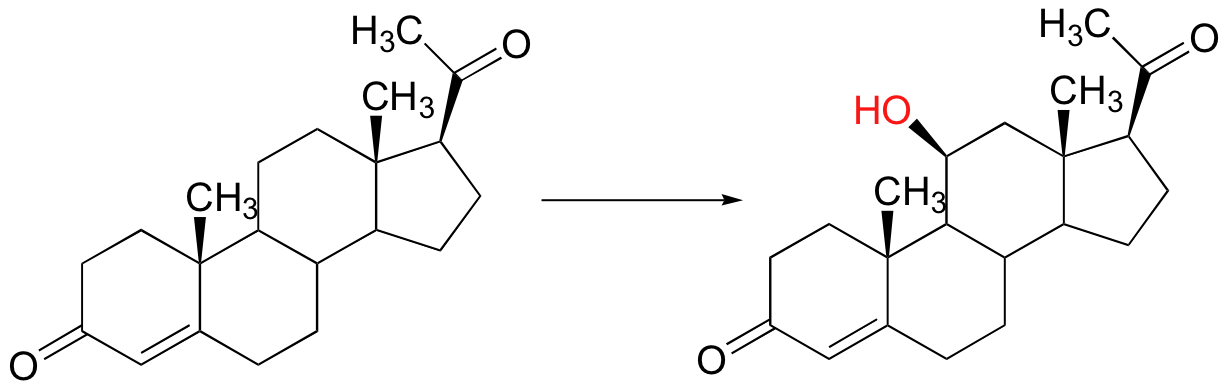
The methylene carbon center involved is simply not reactive enough - there is no acidic proton or electronegative leaving group present, and the carbon is not in any way electrophilic. This alkane hydroxylation, and many others like it, is catalyzed by a family of monooxygenase enzymes, called the 'cytochrome P450' family, that contain a heme redox center. The heme prosthetic group, which you will learn more about in biochemistry and bioinorganic chemistry courses, is characterized by an iron atom coordinated to the nitrogen atoms of four linked pyrrole rings. Two additional sites on the octahedral iron center - at the top and bottom axial positions- are available for coordination, and in the P450 enzymes these sites are occupied by a cysteine sulfur and, after several electron-transfer steps, a neutral oxygen radical atom derived from molecular oxygen.
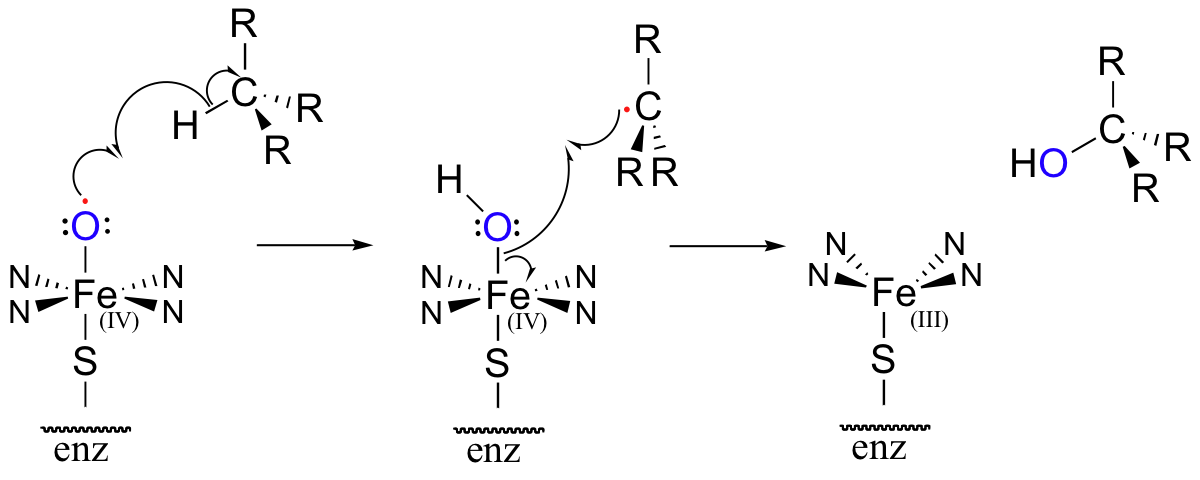
This radical initiator abstracts a hydrogen from an alkane substrate, leading to an alkyl radical intermediate which subsequently recombines with the activated oxygen to form an alcohol and reduced heme (the roman numerals in the figure above refer to the oxidation state of the heme iron – in this process, it gains a single electron and thus is converted from the +4 to the +3 oxidation state).
17.3B: Reductive dehydroxylation of alcohols
In the fatty acid biosynthesis pathway, an alcohol is reduced to an alkane by a two-step dehydration - hydrogenation process (we learned about these reactions in section 16.5). This reductive process somewhat of a special case, however, because the alcohol being reduced is located at the beta position relative to a carbonyl group, and thus enolate intermediates are effectively stabilized.
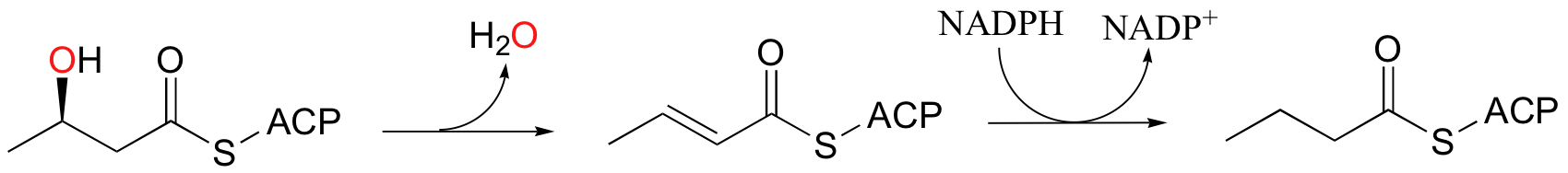
When alcohols which are not located two carbons down from a carbonyl are reduced to alkanes, radical mechanisms are generally involved. One of the most important examples of this type is the reaction catalyzed by ribonucleotide reductase, in which the C2'-hydroxyl group present in RNA nucleotides is removed to form the 2'-deoxynucleotides that make up DNA.
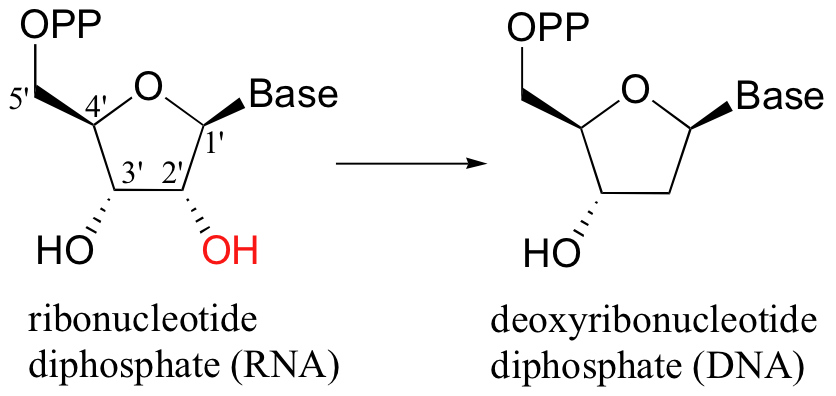
There are several different types of ribonucleotide reductases which are found in different species, but they all appear to operate by the same essential organic mechanism. A radical chain reaction is initiated when an active site cysteine is converted to an unusual thiyl radical - the exact mechanism by which this occurs depends on the type of ribonucleotide reductase enzyme, but all mechanisms are thought to involve the participation of metal redox centers (see Ann. Rev. Biochem 1998, 67, 71 for a detailed review of the various enzyme types). In the interest of clarity, arrows depicting single-electron movement in the mechanism below are colored red, while two-electron arrows are in black.

In the propagation phase of the reaction, the thiyl radical first abstracts a hydrogen from the C3' position of the ribose (step 1), generating an organic radical intermediate. After four more single-electron/two-electron rearrangement steps, the hydroxyl group at C2' is replaced by a hydrogen, a pair of cysteines on the enzyme is oxidized to a disulfide, and the starting thiyl radical is regenerated. To complete the catalytic cycle, the disulfide is reduced to free cysteines by a specific thioredoxin reductase, or by glutathione/glutathione reductase (section 16.12A), depending on the species.
17.3C: Radical mechanisms for flavin-dependent reactions
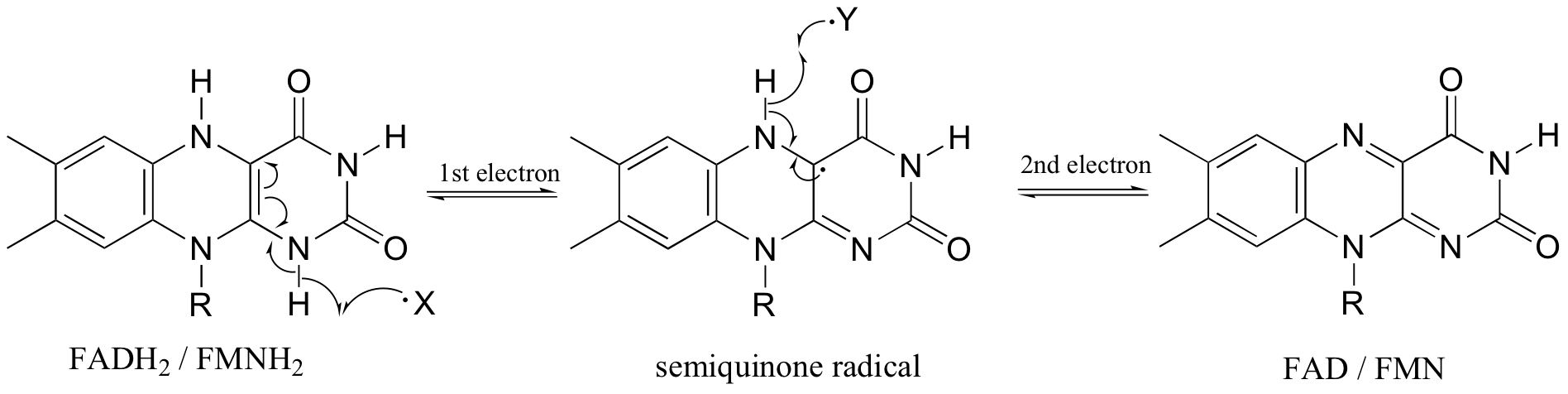
In chapter 16 we saw how flavin coenzymes, like their nicotinamide adenine dinucleotide counterparts, can act as hydride acceptors and donors. In these redox reactions, two electrons are transferred together in the form of a hydride ion. Flavin, however, is also capable of mediating chemical steps in which a single unpaired electron is transferred - in other words, radical chemistry. This is due to the ability of the flavin system to form a stabilized radical intermediate called a semiquinone, formed when FADH2 (or FMNH2) donates a single electron, or when FAD (or FMN) accepts a single electron.
This single-electron transfer capability of flavins is critical to their metabolic role as the entry point of electrons into the electron transport phase of respiration. Electrons 'harvested' from the oxidation of fuel molecules are channeled, one by one, by FMNH2 into the electron transport chain, where they eventually reduce molecular oxygen. NADH is incapable of single electron transfer - all it can do is transfer two electrons, in the form of a hydride, to FMN; the regenerated FMNH2 is then able to continue sending single electrons into the transport chain.
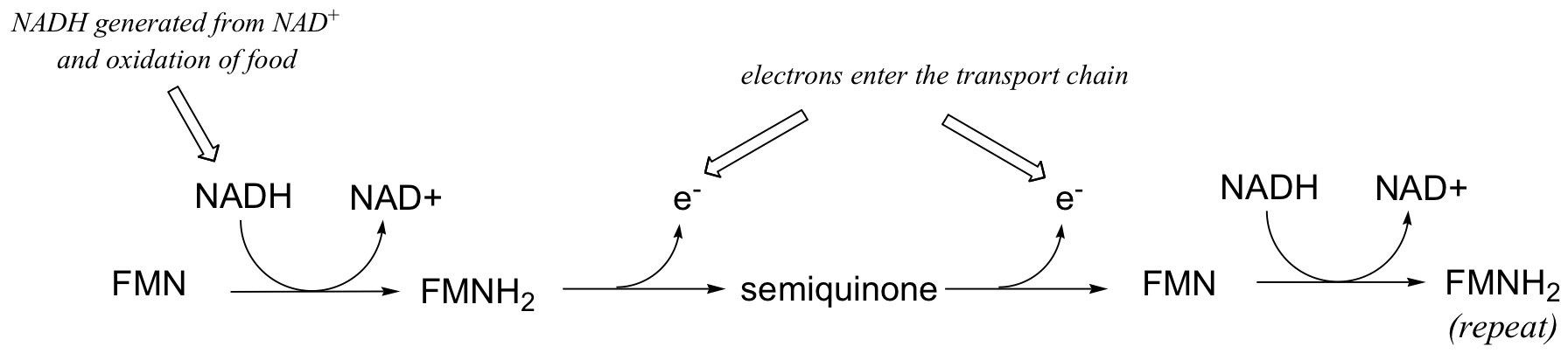
You will learn more details about this process in a biochemistry class.
Because flavins are capable of single-electron as well as two-electron chemistry, the relevant mechanisms of flavoenzyme-catalyzed reactions are often more difficult to determine. Recall the dehydrogenation reaction catalyzed by acyl-CoA dehydrogenase (section 16.5C) - it involves the transfer of two electrons and two protons (ie. a hydrogen molecule) to FAD. Both electrons could be transferred together, with the FAD coenzyme simply acting as a hydride acceptor (this is the mechanism we considered previously). However, because the oxidizing coenzyme being used is FAD rather than NAD+, it is also possible that the reaction could proceed by a single-electron, radical intermediate process. In the alternate radical mechanism proposed below, for example, the enolate intermediate first donates a single electron to FAD, forming a radical semiquinone intermediate (step 2). The second electron is transferred when the semiquinone intermediate abstracts a hydrogen from Cb in a homolytic fashion (step 3).
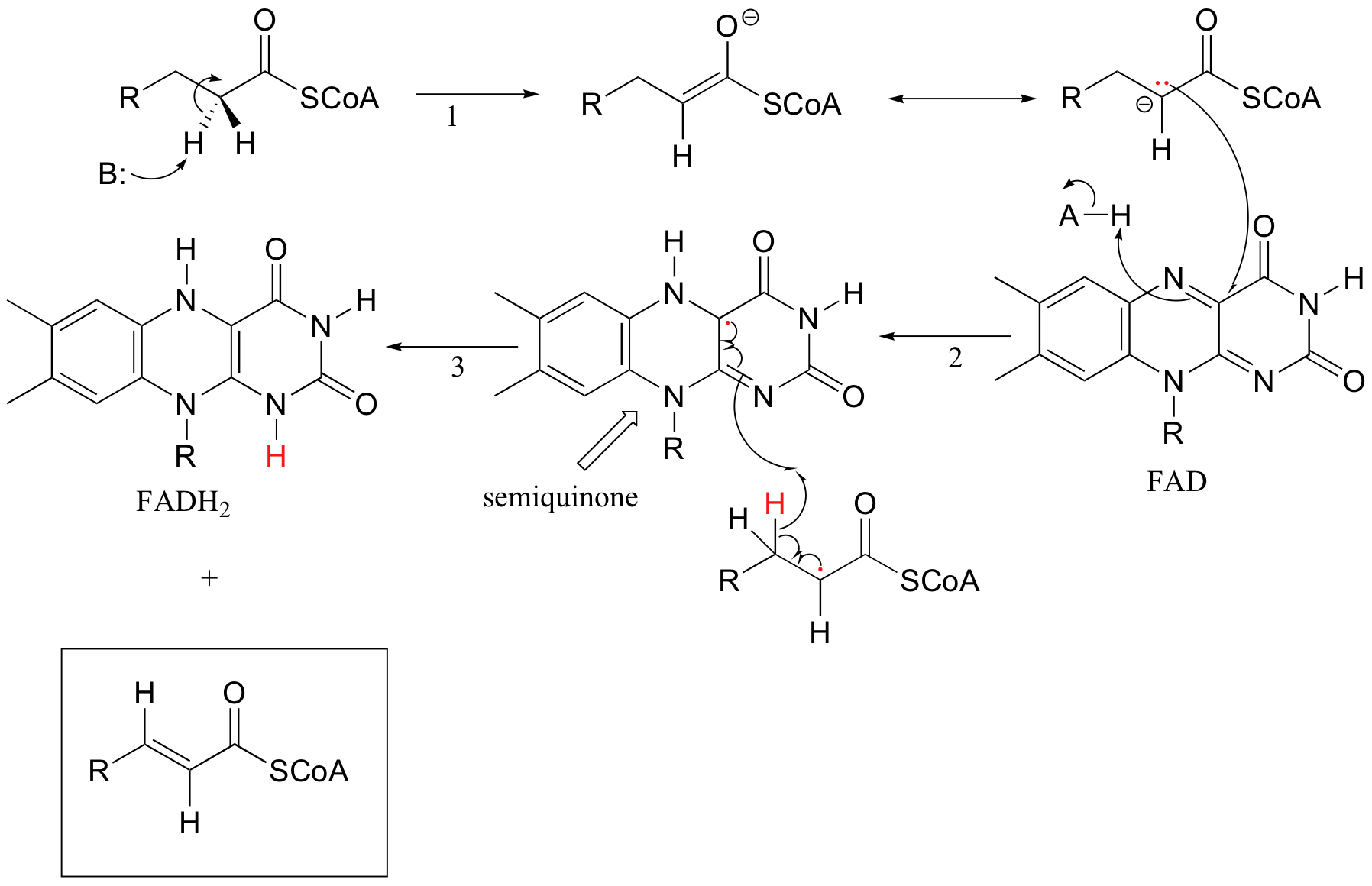
Scientists are still not sure which mechanism - the hydride transfer mechanism that we saw in section 16.5B or the single electron transfer detailed above - more accurately depicts what is going on in this reaction.
The conjugated elimination catalyzed by chorismate synthase (section 14.3B) is another example of a reaction where the participation of flavin throws doubt on the question of what is the relevant mechanism. This could simply be a conjugated E1' reaction, with formation of an allylic carbocation intermediate. The question plaguing researchers studying this enzyme, however, is why FADH2 is required. This is not a redox reaction, and correspondingly, FADH2 is not used up in the course of the transformation - it just needs to be bound in the active site in order for the reaction to proceed. Given that flavins generally participate in single-electron chemistry, this is an indication that radical intermediates may be involved. Recently an alternative mechanism, involving a flavin semiquinone intermediate, has been proposed (J. Biol. Chem 2004, 279, 9451). Notice that a single electron is transferred from substrate to coenzyme in step 2, then transferred back in step 4.
