
16.11: Halogenation of organic compounds
- Page ID
- 1006

16.11A: Enzymatic halogenation
Although naturally occurring halogenated organic compounds are somewhat rare in most animals and plants, the same is not true for many marine organisms, fungi, and bacteria. Natural products chemists have isolated and identified several thousand different naturally occurring halogenated compounds in these species. One important organohalogen natural product that you may have heard of is vancomycin, the current 'antibiotic of last resort' used to treat infections that are resistant to all other antibiotic drugs (increasingly, however, hospitals are encountering infectious bacterial strains that have developed resistance to vancomycin as well). Below is a partial structure of vancomycin showing the two chlorine substituents.
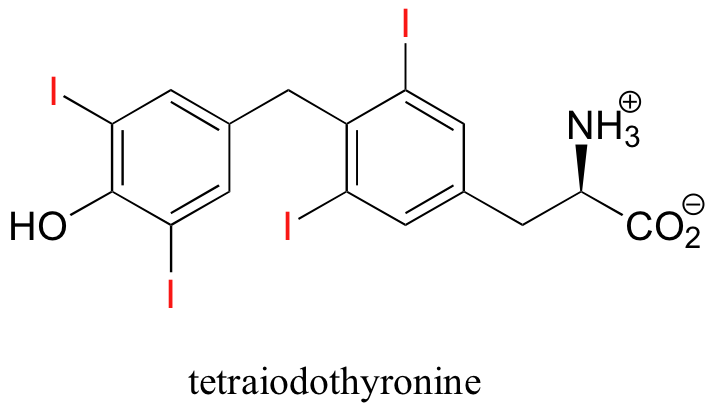
One of the relatively rare examples of an organohalogen compound produced by humans is tetraiodothyronine, an endocrine hormone.
The enzymes which catalyze the incorporation of halogens into organic structures are increasingly becoming a subject of intense interest among enzymologists (see Chemical and Engineering News, May 22, 2006). Because halogens are more electronegative than carbon, halogenations are considered to be redox reactions, and in fact some of the enzymes catalyzing halogenation reactions show strong evolutionary links to oxygenases.
Like the oxygenases, most halogenase enzymes use metal centers - generally iron or vanadium - to accomplish halogen insertion. The basic strategy of these enzymes is to use the metal center to oxidize a halide anion to a cation species (a two-electron oxidation) or radical species (a one-electron oxidation).
\[X^- \rightarrow X^+ + 2e^- \tag{16.11.1}\]
\[X^- \rightarrow X^• + e^- \tag{16.11.2}\]
The halogen cations can then react as electrophiles with alkenes and aromatic rings, while the radical halogen intermediates go on to halogenate unactivated alkyl groups (alkane hydroxylation reactions also proceed through radical intermediates – radical hydroxyation and halogenation reactions are discussed in more detail in chapter 17).
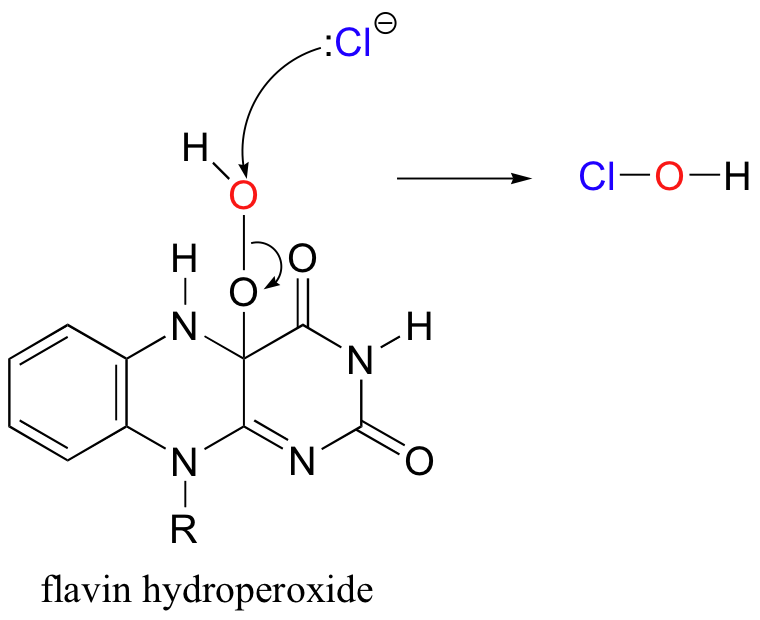
Recently, a metal-free halogenase was discovered that uses only FADH2 to introduce a chlorine to the aromatic ring of tryptophan (Science 2005, 309, 2216). The reaction mechanism relies on the generation of flavin hydroperoxide from FADH2 and molecular oxygen, just like the two flavin-dependant monooxygenases discussed earlier. Chloride ion attacks the hydroperoxide, generating ClOH, or hypochlorous acid - essentially the same as household bleach!
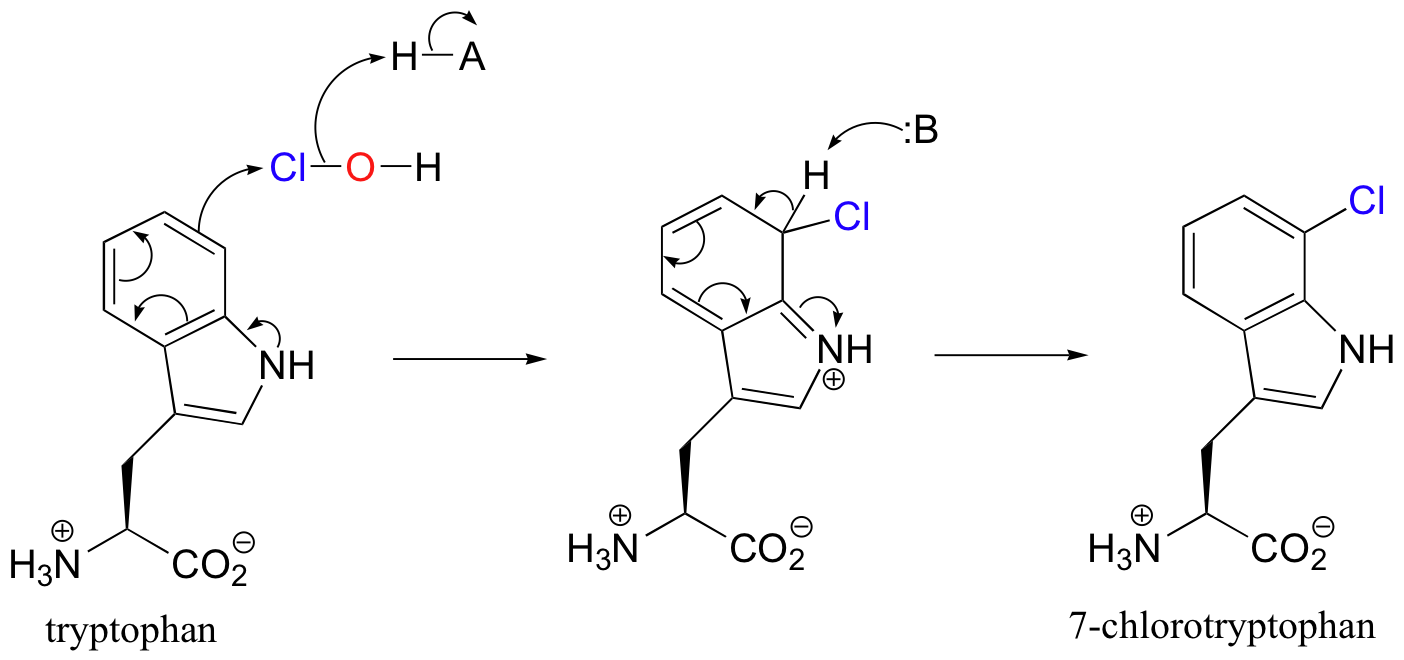
After traveling through a 10 angstrom long tunnel inside the enzyme, the HOCl intermediate comes into contact with C7 of tryptophan, and reacts as the electrophilic partner in an electrophilic aromatic substitution.
Although detailed discussion is beyond the scope of this text, it is worth mentioning that many microbes have enzymes with the specific function of removing halogen atoms from organic compounds (this of course is a reductive transformation). These enzymes are the subject of intense interest as potential bioremediation tools to detoxify halogenated industrial pollutants that have been released into the environment (see Environ. Microbiol. 2005, 7, 1868 for a review).
16.11B: Synthetic parallel - halogenation of alkenes in the lab
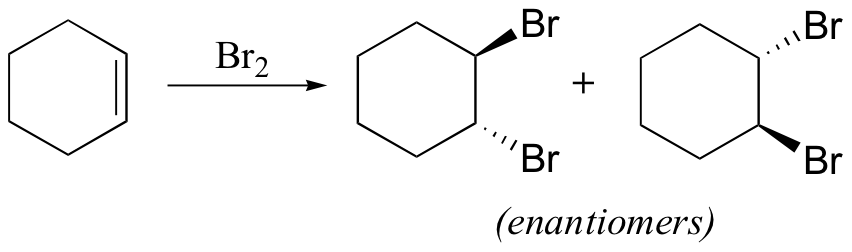
The enzymatic halogenation reactions discussed here have parallels in laboratory synthesis. When an alkene is treated with a solution of Br2 in organic solvent, the bromine adds in a trans fashion to the double bond, via a bromonium ion intermediate.
How is molecular bromine an electrophile? Because of the size of the two bromine atoms in Br2, the electrons in the molecule are very polarizable, meaning that they can be easily 'pulled' toward an electron-poor species, or 'pushed' away from an electron-rich species. The double bond of the alkene is, of course, electron-rich, due to the electrons in the π bond. When the Br2 molecule approaches one face of the double bond, the π electron density 'pushes' electron density in the Br2 molecule away from the closer Br atom, giving it a partial positive charge, and towards the more distant Br atom, giving it a partial negative charge.

This is called an 'induced dipole'. The closer Br atom thus becomes electrophilic, and is attacked by the π electrons of the alkene (this is an electrophilic step, like those we learned about in chapter 15).
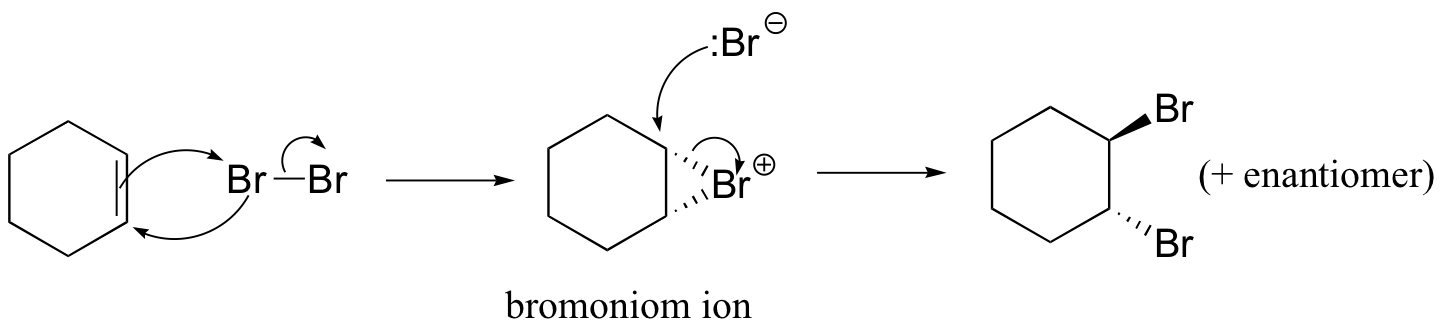
Because the Br-Br s bond is relatively weak, the second Br atom is expelled as bromide anion, and a cyclic organobromonium ion is formed. The bromine anion then attacks one of the carbons in the organobromoniom ion from the backside, opening the ring and completing the trans addition process.
The 'bromine test' is a quick and convenient way to check for the presence of carbon-carbon double bonds. A Br2 solution has a distinctive dark red-brown color that disappears rapidly when the molecular bromine adds to a double bond to form a (colorless) organic dibromide. Cyclohexane and cyclohexene can easily be distinguished, for example, by adding a few drops of bromine solution to a sample of each. The red-brown color of the bromine will immediately disappear in the cyclohexene sample as it adds to the double bond, but will linger in the cyclohexane sample. This type of qualitative test for the presence of a particular functional group was widely used before the development of IR spectroscopy.
Several mechanistically similar variations on the dibromination reactions described above are also possible. If bromine is added to an alkene in the presence of water, for example, a "halohydrin" compound will form. If saturated NaCl solution is used, some 1-bromo-2-chloro product will result.
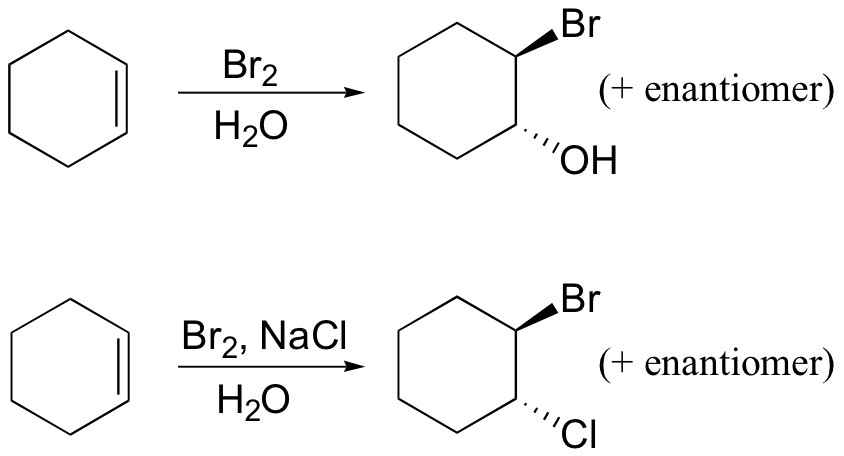
Example 16.8
Draw mechanisms for the halogenation reactions shown above.
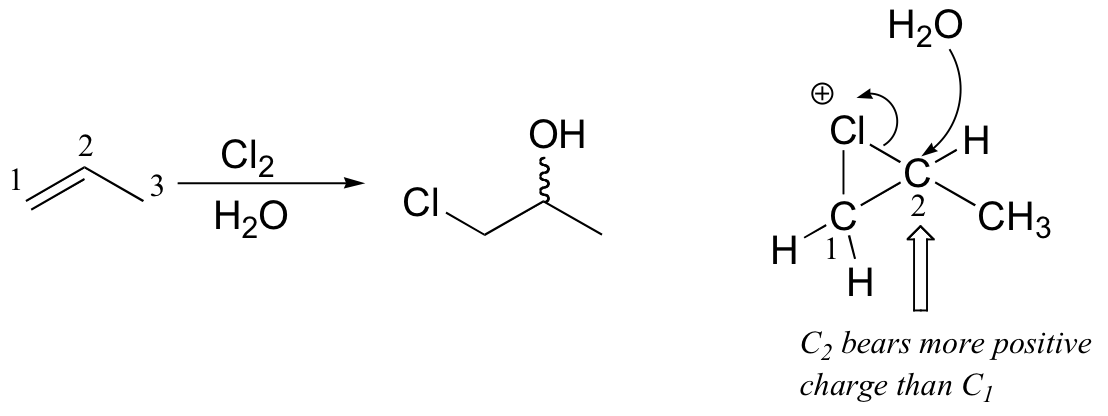
This is because, in the chloronium ion intermediate, C2 (a secondary carbon) bears more positive charge than C1, and thus is attacked preferentially by the water nucleophile (review relative carbocation stabilities in section 8.4B).