
20.1: Oxidation-Reduction Reactions of Organic Compounds- An Overview
- Page ID
- 13991
Synthetic organic chemists have a wide range of reagents at their disposal for the reduction or oxidation of functional groups in organic compounds. The reagent to be used for any given transformation must be chosen carefully in order to ensure that only the desired functional group or groups is effected: some reducing agents, for example, will act on ketones and aldehydes but leave alkenes and carboxylic acid derivatives untouched, while other will reduce all of these functional groups. Different redox reagents will also transform groups to different extents: we will soon see oxidizing agents, for example, that will transform a primary alcohol to a carboxylic acid, and others that, given the same primary alcohol, will produce an aldehyde. Similarly, reduction of an alkyne can produce a cis-alkene, a trans-alkene, or an alkane, depending on the reducing agent used. In this section, we will take a look at the action of some of the most important redox reactions – those that are used most frequently in the laboratory, and those which, perhaps more importantly for some of you, tend to make their appearance on standardized tests such as the MCAT. A much more complete discussion of redox reagents can be found in advanced organic synthesis textbooks and reference sources. It also important to bear in mind that increasingly, synthetic organic chemists are figuring out how to use redox enzymes as tools to catalyze the reactions that they wish to carry out in the lab (Curr. Opin. Biotechnol. 2003, 14, 427; Adv. Biochem. Eng. Biotechnol. 2005, 92, 261).
16.13A: Metal hydride reducing agents
In the organic synthesis laboratory, carbonyl groups can be reduced using hydride transfer reactions that are mechanistically similar to biochemical reactions with NAD(P)H. Three common reducing agents are sodium borohydride (NaBH4), lithium aluminum hydride (LiAlH4), and diisobutyl aluminum hydride (DIBAH).

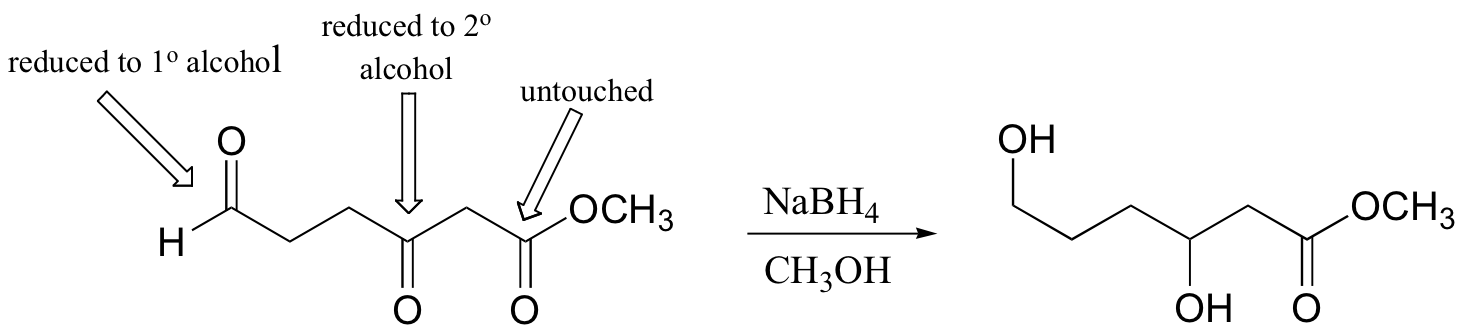
For example, when sodium borohydride is stirred in solution with an aldehyde or ketone, a hydride ion adds to the carbonyl carbon to form a 2o alcohol (from a ketone) or a 1o alcohol (from an aldehyde).
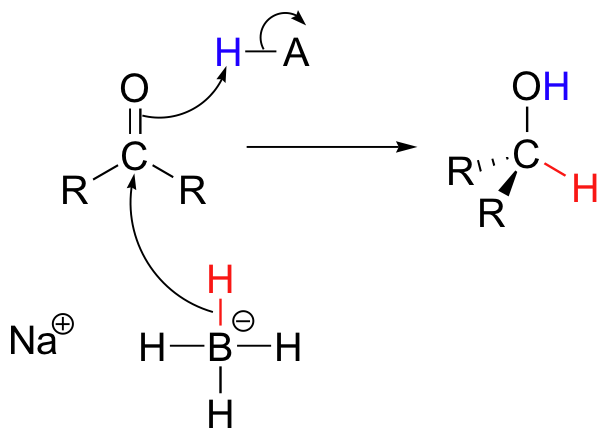
Sodium borohydride is a relatively mild reducing agent, and reactions are typically run in water, methanol, or ethanol solvent. One mole of NaBH4is capable of reducing four moles of ketone or aldehyde. Carboxylic acid derivatives and alkene double bounds are not affected.
LiAlH4 works in a manner similar to NaBH4, but is much more reactive. It will react violently with protic solvents (like water or methanol), and so an organic solvent such as diethyl ether must be used. LiAlH4 will not affect alkene double bonds, but unlike NaBH4 it will reduce carboxylic acids and esters (to 1o alcohols), amides (to amines), nitriles (to 1oamines), and can even be used in reductive ring-opening reactions with epoxides to form alcohols.
DIBAH has only one hydride to deliver (as opposed to four for NABH4 and LiAlH4), and if only one molar equivalent is used it can reduce an ester to an aldehyde. Using LiAlH4 would reduce the ester to a primary alcohol, as would using two molar equivalents of DIBAH.
In all of these metal hydride reductions, hydride addition can occur from either side of the carbonyl, meaning that reduction of an asymmetrical ketone will result in a racemic mix of both R and S alcohols. For most ketones, this mixture will be present at a ratio of approximately 50:50, because the likelihood of hydride attack is equal at either side. In some cases, however, the two faces presented by the ketone group are not equivalent. Camphor, a natural compound with a distinctive smell that is used in many cosmetics and home health products, is a case in point. Looking at the structure of the camphor molecule, you can see that, because of the conformational rigidity of the fused ring structure, the re and si faces of the carbonyl group are not equivalent - approach by a nucleophile appears to be less hindered from the bottom (si) side than from the top (re) side, (it is easier to visualize this if you build a model).

This can be confirmed by analyzing the 1H NMR spectrum of the purified product from a reaction with NaBH4. This spectrum is actually quite complex, because it is not a pure sample but a mixture of two different diastereomers - there are many overlapping peaks and complex splitting patterns that are difficult to interpret. Fortunately, however, the signals corresponding to the hydrogen atom of interest (HT and HB for the products of topside and bottomside attack, respectively) are quite distinct - they are both in a region of the spectrum devoid of any other peaks. HT in one diastereomer has a chemical shift of 4.0 ppm, while HB in the other diastereomer has a chemical shift of 3.6 ppm. The integration value of these two peaks relative to one another is of course equal to the ratio of the two diastereomers in the product mixture and, as expected, integration shows that there is substantially more of the product that results from bottomside attack. (Pavia, et al., "Introduction to Organic Laboratory Techniques - A Contemporary Approach", 2nd ed. 1982, CBS College Publishing, Philadelphia USA).
16.13B: Catalytic hydrogenation and the trans fat issue
In section 16.5, we saw several examples of enzymatic alkene hydrogenation reactions. In the organic chemistry lab, hydrogenation of alkenes is generally carried out with hydrogen gas on the surface of a metal catalyst such as platinum, palladium, or nickel. This process is usually referred to as catalytic hydrogenation. Although the exact mechanism by which the reaction occurs is still a topic of debate, the general picture is fairly clear. Empty orbitals on the metal allow for hydrogen to be adsorbed on the catalytic surface. The alkene also complexes to the metal surface, as the π electrons in the double bond also interact with empty metal orbitals. At this point, hydrogen is inserted into the double bond, and the reduced alkane product is released.
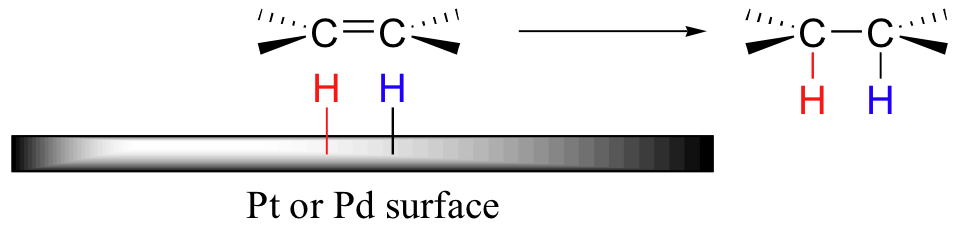
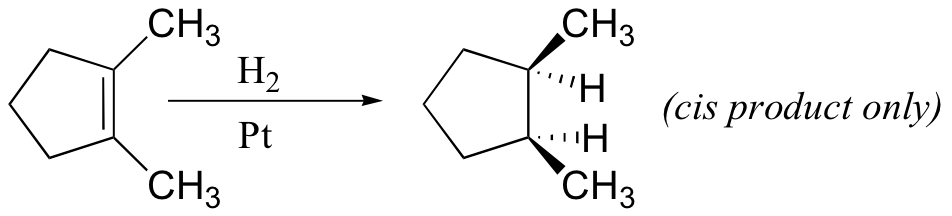
Because this reaction takes place on a planar surface, addition of hydrogen occurs on the same face of the double bond - a syn addition, in other words. The catalytic hydrogenation of 1,2-dimethylcyclopentane will yield, for example, the cis dimethylcycloalkane product, with little or no formation of a trans product.
It is not only alkene double bonds that are reduced by catalytic hydrogenation: alkynes are reduced to alkanes, aldehydes and ketones are reduced to their corresponding alcohols, and nitro groups are reduced to amines. Carboxylic acid derivatives, however, are not affected, and aromatic double bonds are also left untouched.
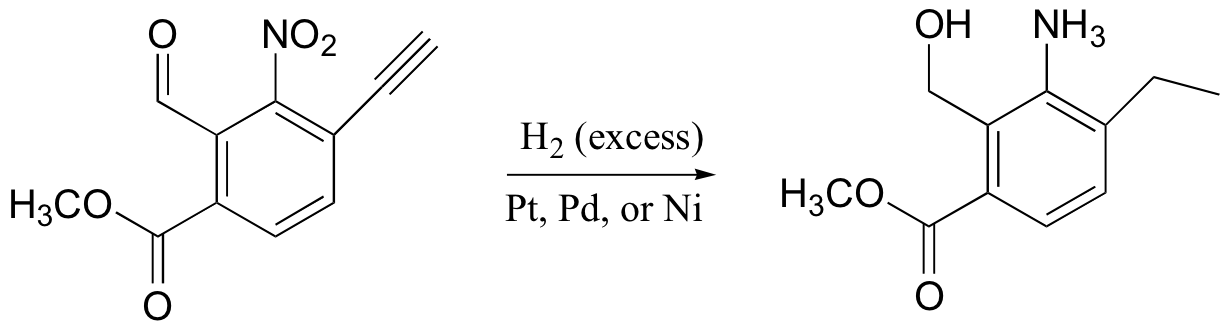
Catalytic hydrogenation of alkenes is currently a hot topic in food chemistry. Margarine is produced by partial hydrogenation of double bonds in the unsaturated fatty acids in liquid vegetable oils, usually with a nickel catalyst. Complete hydrogenation would produce fully saturated fatty acids and lead to a lard-like product that is too hard to spread on toast, so conditions are adjusted to ensure that only some of the double bonds are hydrogenated while others are left in place, resulting in a soft and spreadable product. This process is called partial hydrogenation. (review the chemical basis of the relationship between lipid saturation and melting point in section 2.4D).
Recently, however, scientists have become increasingly worried about the presence of unnatural fatty acids found in margarine and other food products made from partially hydrogenized oils. Natural unsaturated fatty acids have mainly cis double bonds. In the unnatural fatty acids found in margarines, the naturally-occurring cis stereochemistry has been converted to trans. Trans-fatty acids have been associated with heart disease and some forms of cancer.
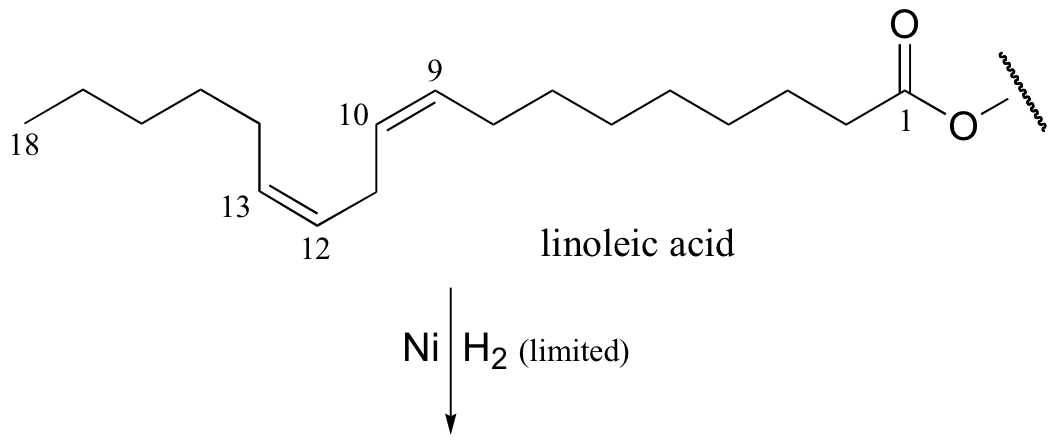
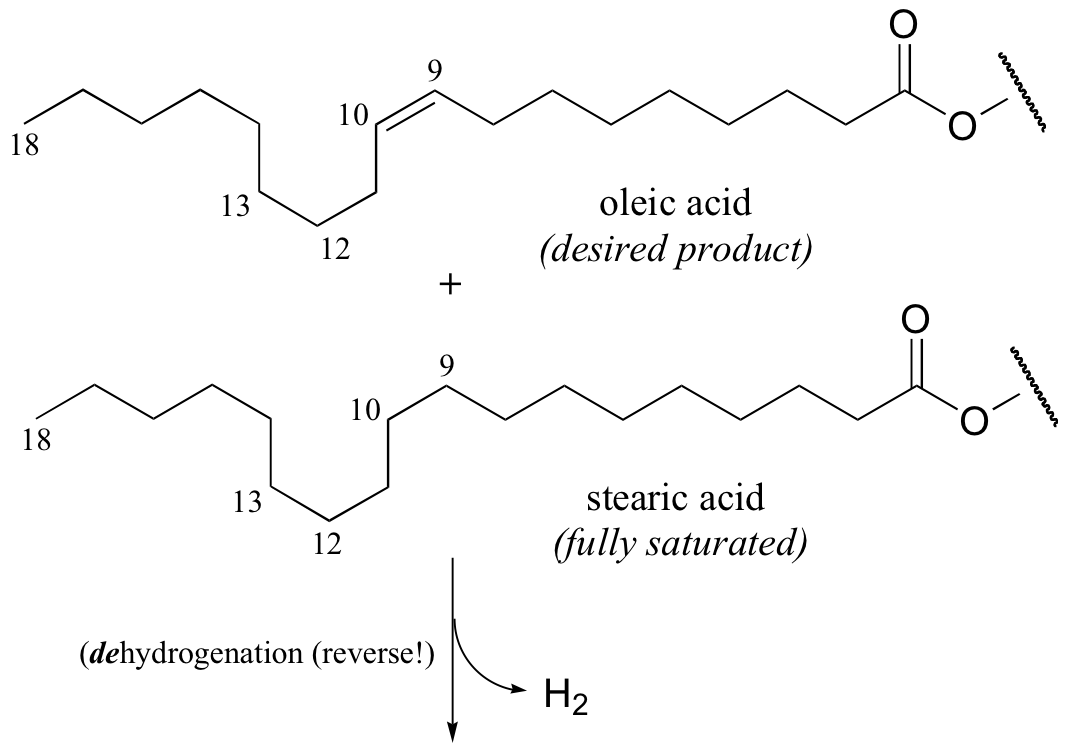
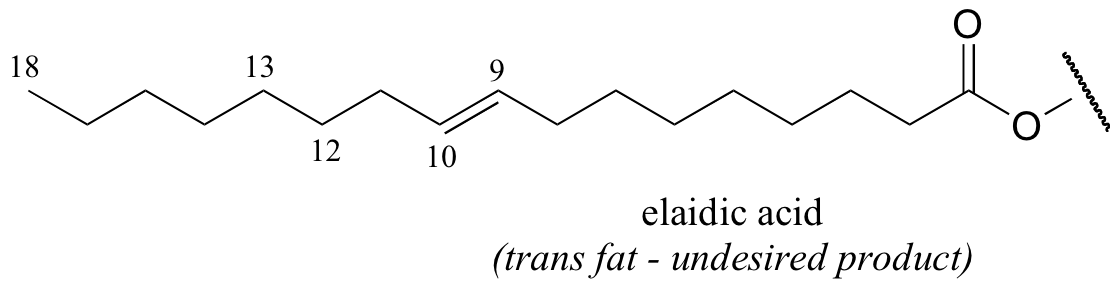
It appears that these unnatural trans fatty acid isomers are unintentionally produced by the hydrogenation process. The problem is that there is no control over the regiochemistry or stereochemistry of the reverse (dehydrogenation) reaction. Because only a limited amount of hydrogen is used in order to achieve partial (rather then complete) hydrogenation, the process is reversible, meaning that double bonds tend to re-form - and when they do, it is often in the lower-energy trans configuration, rather than the natural cis configuration. The figure below shows the partial hydrogenation of a linoleic acid hydrocarbon over a nickel catalyst, resulting in oleic acid, which is the desired cis-unsaturated product, as well as elaidic acid, the undesirable trans fat product.
Food producers are increasingly adopting alternative hydrogenation technologies and production strategies in order to offer products that are free of trans fatty acids. (See J. Am. Diet. Assoc. 2006, 106, 867 for a detailed review of this topic).
What if we want to convert the triple bond of an alkyne to the double bond of an alkene, rather than all the way to an alkane single bond? Fortunately, there is a way to do this – an even better, we can choose whether to make a cis or a trans double bond! To convert an alkyne to a cis-alkene, we carry out catalytic hydrogenation reaction using what is known as a ‘Lindlar catalyst" – finely powdered palladium depositied on calcium carbonate and modified with lead salts. This is essentially a less reactive version of the normal transition metal catalyst used in hydrogenation of alkenes.
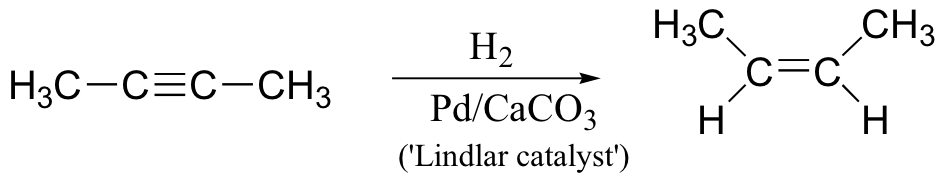
If we want to make a trans alkene, we use sodium metal and liquid ammonia in a very different type of reaction – the ‘dissolving metal reduction’:
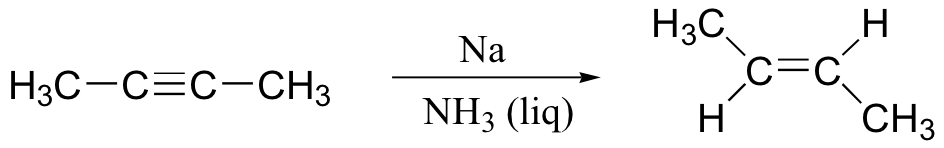
This reaction mechanism is thought to occur through radical intermediates, but we will not concern ourselves with exactly how it works.
16.13C: Reduction of carbonyl carbons to methylene
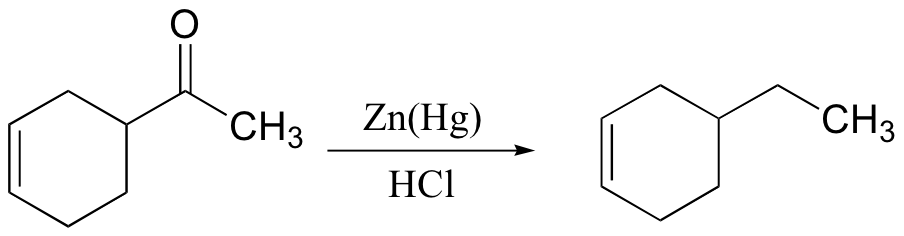
There are two principle methods for reducing the carbonyl group of a ketone to a simple methylene (CH2) carbon. The mechanism for the Clemmensen reduction is not well understood, but you will be asked to propose a mechanism for the Wolff-Kishner reduction in the end-of-chapter problems.
The Clemmensen reduction:
The Wolff-Kishner reduction:
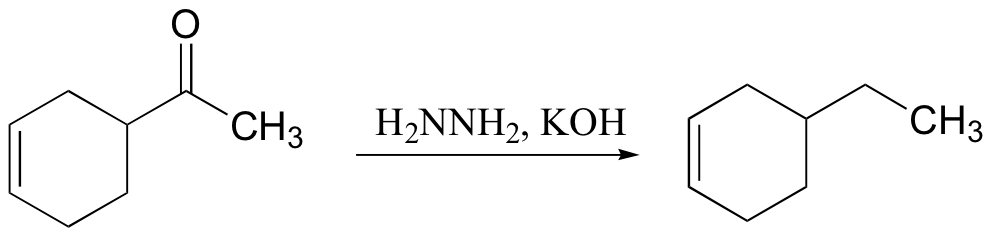
16.13D: Laboratory oxidation reactions
The laboratory oxidation of an alcohol to form an aldehyde or ketone is mechanistically different from the biochemical oxidations with NAD(P)+ that we saw earlier in this chapter. The general picture of laboratory oxidations is illustrated below. Essentially what happens is that the hydroxide hydrogen of the alcohol is replaced by a leaving group (X in the figure below).
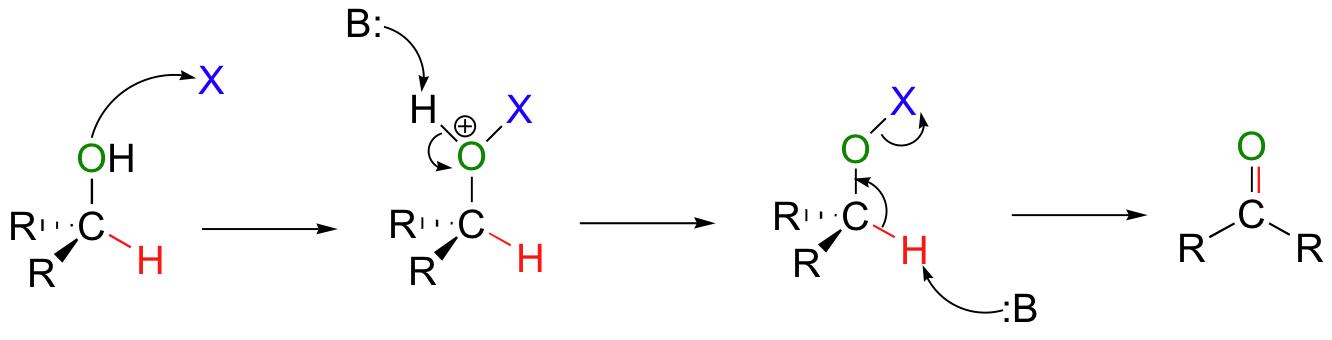
Then, a base can abstract the proton bound to the alcohol carbon, which results in elimination of the X leaving group and formation of a new carbon-oxygen double bond. As you can see by looking closely at this general mechanism, tertiary alcohols cannot be oxidized in this way – there is no hydrogen to abstract in the final step!
A common method for oxidizing secondary alcohols to ketones uses chromic acid (H2CrO4) as the oxidizing agent. Chromic acid, also known as Jones reagent, is prepared by adding chromium trioxide (CrO3) to aqueous sulfuric acid.
A mechanism for the chromic acid oxidation of a ketone is shown below.
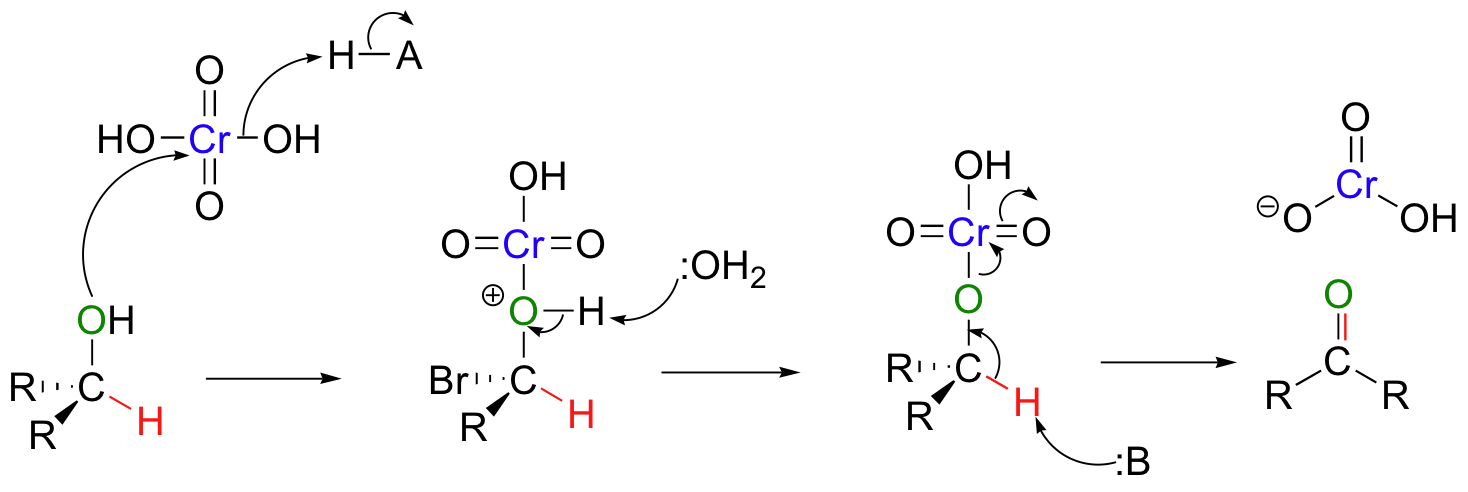
Note that the chromium reagent has lost two bonds to oxygen in this reaction, and thus has been reduced (it must have been reduced - it is the oxidizing agent!).
Ketones are not oxidized by chromic acid, so the reaction stops at the ketone stage. In contrast, primary alcohols are oxidized by chromic acid first to aldehydes, then straight on to carboxylic acids.
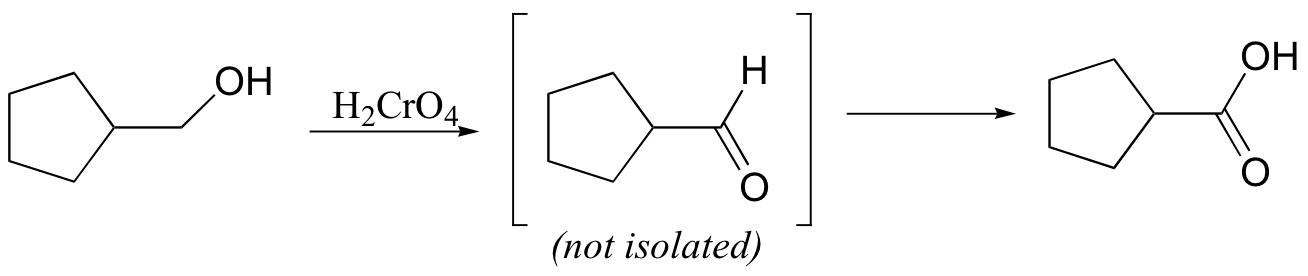
It is actually the hydride form of the aldehyde that is oxidized (recall from section 11.3 that aldehydes in aqueous solution exist in rapid equilibrium with their hydrate forms).
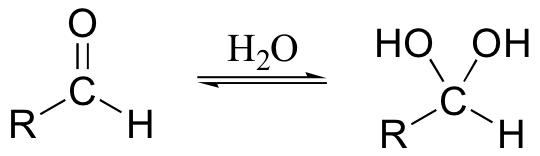
One of the hydroxyl groups of the hydrate attacks chromic acid, and the reaction proceeds essentially as shown for the oxidation of a secondary alcohol.
Under some conditions, chromic acid will even oxidize a carbon in the benzylic position to a carboxylic acid (notice that a carbon-carbon bond is broken in this transformation).
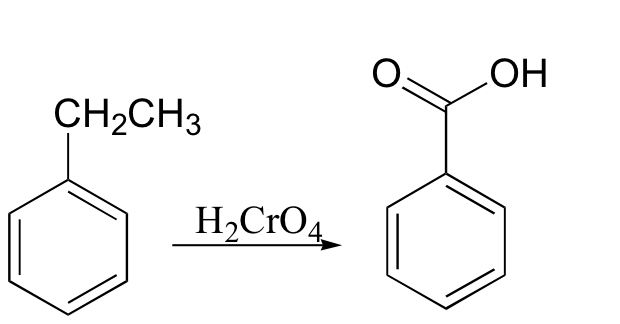
A number of other common oxidizing agents are discussed below.
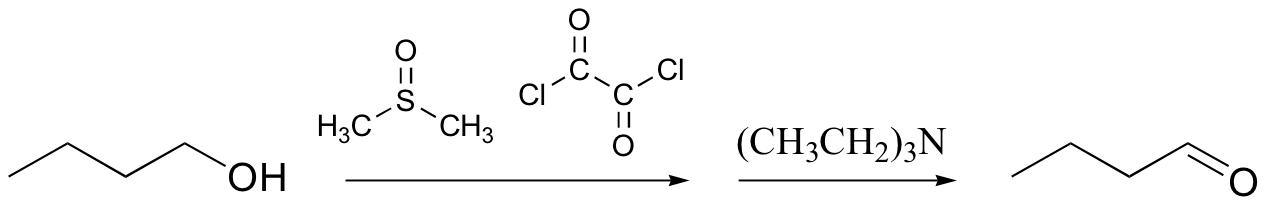
The pyridinium chlorochromate (PCC) and Swern oxidation reactions are useful for oxidizing primary alcohols to aldehydes. Further oxidation of the aldehyde to the carboxylic acid stage does not occur with these reagents, because the reactions are carried out in anhydrous (water-free) organic solvents such as dichloromethane, and therefore the hydrate form of the aldehyde is not able to form.
The Swern oxidation uses dimethylsulfoxide and oxalyl chloride, followed by addition of a base such as triethylamine. The actual oxidizing species in this reaction is the dimethylchlorosulfonium ion, which forms from dimethylsulfoxide and oxalyl chloride.
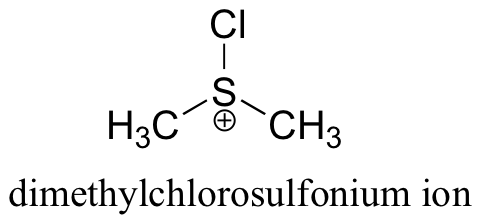
You will be asked to propose a mechanism for these reactions in the end of chapter problems.
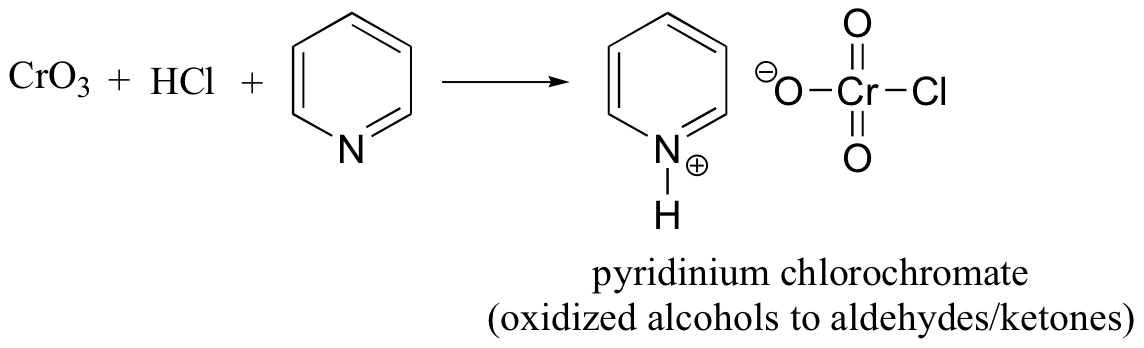
Pyridinium chlorochromate is generated by combining chromium trioxide, hydrochloric acid, and pyridine.
The PCC and Swern oxidation conditions can both also be used to oxidize secondary alcohols to ketones.
Silver ion, Ag(I), is often used to oxidize aldehydes to ketones. Two common reaction conditions are:
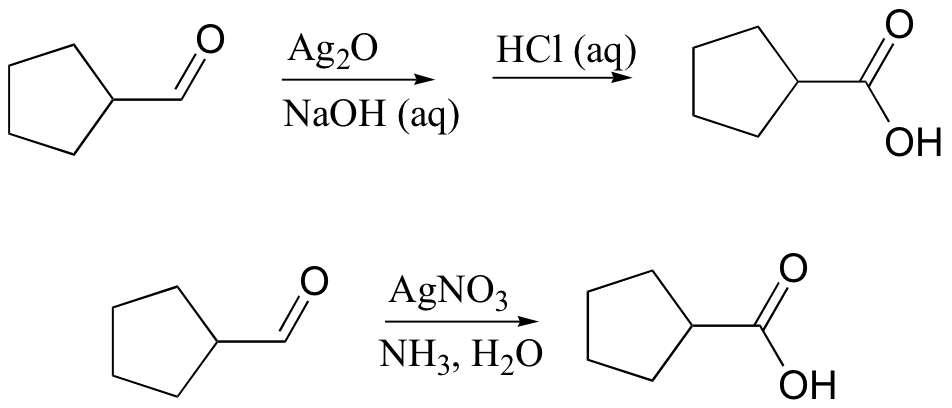
The set of reagents in the latter reaction conditions are commonly known as ‘Tollens’ reagent’.
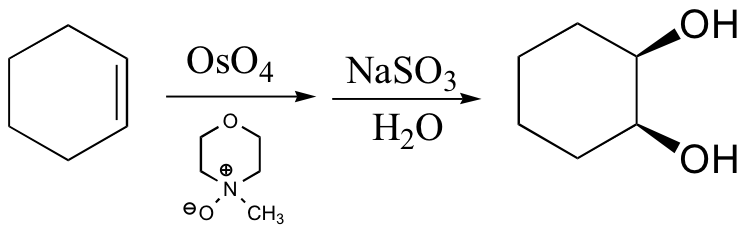
Alkenes are oxidized to cis-1,2-diols by osmium tetroxide (OsO4). The stereospecificity is due to the formation of a cyclic osmate ester intermediate. Osmium tetroxide is used in catalytic amounts, and is regenerated by N-methylmorpholine-N-oxide.
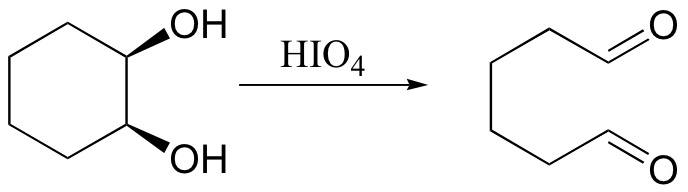
cis-1,2-diol compounds can be oxidized to dialdehydes (or diketones, depending on the substitution of the starting diol) using periodic acid:
Alkenes can also be oxidized by treatment with ozone, O3. In ozonolysis, the carbon-carbon double bond is cleaved, and the alkene carbons are converted to aldehydes:
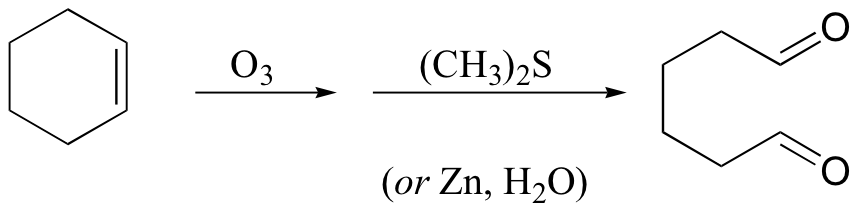
Dimethyl sulfide or zinc is added in the work-up stage of the reaction in order to reduce hydrogen peroxide, which is formed in the reaction, to water.
Alternatively, hydrogen peroxide and aqueous base can be added in the workup to obtain carboxylic acids:
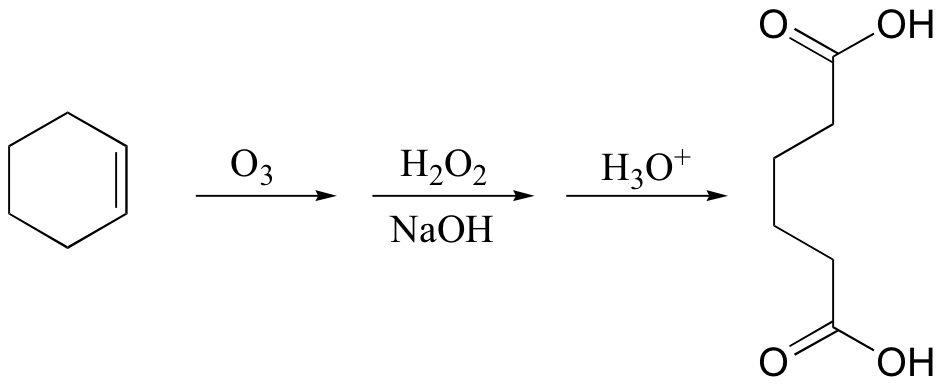
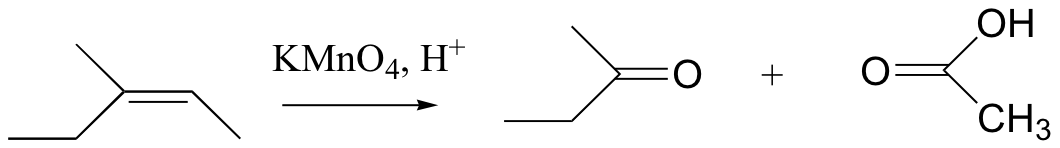
Potassium permanganate (KMnO4) is another very powerful oxidizing agent that will oxidize primary alcohols and aldehydes to carboxylic acids. KMnO4 is also useful for oxidative cleavage of alkenes to ketones and carboxylic acids:
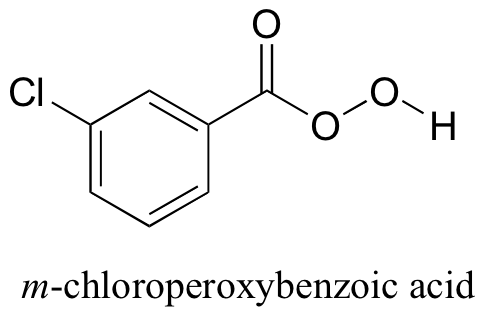
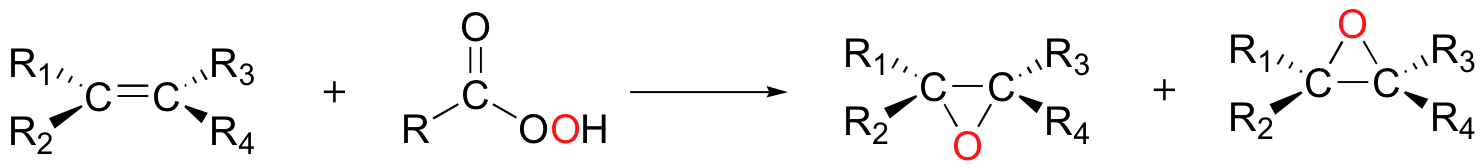
Finally, alkenes can be oxidized to epoxides using a 'peroxyacid' such as m-chloroperoxybenzoic acid. Notice the presence of a third oxygen in the peroxyacid functional group.
The mechanism is similar to that of the biological epoxidation catalyzed by squalene epoxidase (section 16.10A), with the π electrons in the alkene double bond attacking the 'outer' oxygen of the peroxyacid and cleaving the reactive O-O peroxide bond.
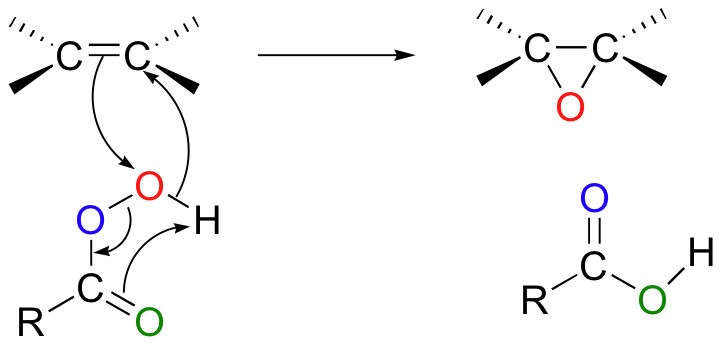
Uncatalyzed epoxidation of an asymmetric alkene generally results in two diastereomeric epoxide products, with the epoxide adding either from above or below the plane of the alkene.
Epoxides are very useful intermediates in organic synthesis. Because most naturally occurring molecules (including those with medicinal properties) are chiral, control of stereochemistry is one of the most important challenges facing a synthetic chemist attempting to synthesize a naturally occurring molecule in the laboratory. In what was arguably one of the most important discoveries in synthetic organic chemistry in recent decades, Barry Sharpless of Stanford University reported in 1980 that he and his colleagues had developed a method to stereoselectively epoxidize asymmetric alkenes which contained an alcohol in the allylic position. The ‘Sharpless asymmetric oxidation’ is achieved with the use of a chiral catalyst composed of (+) or (-) diethyltartrate and an organotitanium compound (J. Am. Chem. Soc. 1980, 102, 5974). Depending on which stereoisomer of diethyltartrate is used, the peroxyacid oxygen tends to add to either the top or bottom plane of the alkene.

This technique allows for the specific introduction of two new stereocenters at an alkene position, which as you can imagine makes it an extremely useful synthetic tool.