
17.7: Experimental methods of chemical kinetics
- Page ID
- 46074

Studies of the dynamics of chemical processes impinge on almost every area of chemistry and biochemistry. It is useful for students even at the general chemistry level to have some understanding of the experimental techniques that have informed what you have already learned about kinetics.
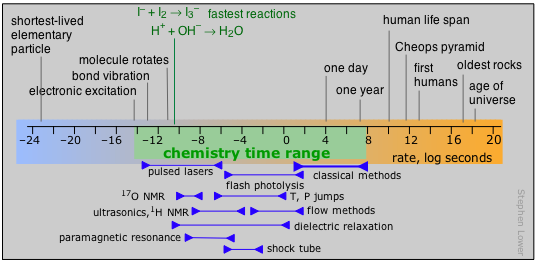
The time domain of chemical kinetics
Kinetics deals with rates of change, so as a preface to this section, it is useful to consider the range of times within which the study of chemical processes takes place. Prior to about 1945, the practical range for kinetic investigations began at about 103 sec and extended out to around 108 sec, this upper limit being governed by the length of time that a graduate student (who usually does the actual work) can expect to get a Ph.D. degree. The development of flash photolysis and flow methods in the mid-1940s moved the lower time limit down to the millisecond range. The most dramatic changes began to occur in the 1970s as the availablity of lasers capable of very short-lived pulses and fast electronics gradually opened up the study of reactions that are completed in nanoseconds, picoseconds, and even femtoseconds (10–15 sec).
The basic way of obtaining the information needed to determine rate constants and reaction orders is to bring the reactants together and then measure successive changes in concentration of one of the components as a function of time. Two important requirements are
- The time required to take a measurement must be very short compared to the time the reaction takes to run to completion;
- The temperature must be held constant — something than can pose a problem if the reaction is highly exothermic.
Measuring the concentration of a reactant or product directly — that is by chemical analysis, is awkward and seldom necessary. When it cannot be avoided, the reaction sample must usually be quenched in some way in order to stop any further change until its composition can be analyzed. This may be accomplished in various ways, depending on the particular reaction. For reactions carried out in solution, especially enzyme-catalyzed ones, it is sometimes practical to add a known quantity of acid or base to change the pH, or to add some other inhibitory agent. More commonly, however, the preferred approach is to observe some physical property whose magnitude is proportional to the extent of the reaction.
Classical and laboratory-class methods
These methods are applicable to reactions that are not excessively fast, typically requiring a few minutes or hours to run to completion. They were about the only methods available before 1950. Most first-year laboratory courses students will include at least one experiment based on one of these methods.
Optical methods
Light absorption is perhaps the most widely-used technique. If either a reactant or a product is colored, the reaction is easily followed by recording the change in transmission of an appropriate wavelength after the reaction is started.
When a beam of light passes through a solution containing a colored substance, the fraction that is absorbed is directly proportional to the concentration of that substance and to the length of the light's path through the solution. The latter can be controlled by employing a cell or cuvette having a fixed path length.
If I0 is the intensity of the light incident on the cell and I is the intensity that emerges on the other side, then the percent absorption is just 100 × I / I0. Because a limited 1-100 scale of light absorption is often inadequate to express the many orders of magnitude frequently encountered, a logarithmic term optical density is often employed. The relation between this, the cell path length, concentration, and innate absorption ability of the colored substance is expressed by Beer's law.
The simplest absorbance measuring device is a colorimeter in which a beam of white light from an incandescent lamp is passed through a cell (often just a test tube) and onto a photodetector whose electrical output is directly proportional to the light intensity. Before beginning the experiment, a zeroing control sets the meter to zero when the light path is blocked off, and a sensitivity control sets it to 100 when a cell containing an uncolored solution (a "blank") is inserted.
The sensitivity and selectivity of such an arrangement is greatly enhanced by adjusting the wavelength of the light to match the absorption spectrum of the substance being measured. Thus if the substance has a yellow color, it is because blue light is being absorbed, so a blue color filter is placed in the light path.
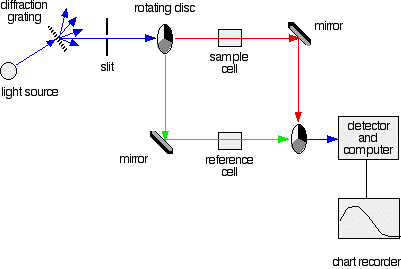
More sophisticated absorption spectrophotometers employ two cells (one for the sample and another reference cell for the blank. They also allow one to select the particular wavelength range that is most strongly absorbed by the substance under investigation, which can often extend into the near-ultraviolet region.
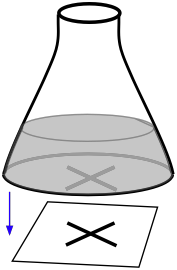
Light scattering measurements (made with a nephthelometer) can be useful for reactions that lead to the formation a fine precipitate/ A very simple student laboratory experiment of this kind can be carried out by placing a conical flask containing the reaction mixture on top of a marked piece of paper. The effects of changing the temperature or reactant concentrations can be made by observing how long it takes for precipitate formation to obscure the mark on the paper.
Other optical methods such a fluorescence and polarimetry (measurement of the degree to which a solution rotates the plane of polarized light) are also employed when applicable.
Other methods
- Measurement of gases: The classical method of following reactions that produce changes in the number of moles of gases is to observe changes in pressure or volume. An alternative method for following the loss of a gas is to place the reaction container on an electronic balance and monitor the loss in weight. This is generally less accurate than pressure measurements, but is sometimes used in student experiments.
- pH measurements: Many reactions yield or consume hydrogen ions and are conveniently followed by means of a pH meter.
- Electrical Conductance: Reactions that yield or consume ionic substances are often studied by measuring the electrical conductance of the solution. Conductimetry is usually carried out by balancing the solution conductance with a known resistance in a bridge arrangement. An audio-frequency alternating current is used in order to avoid electrolysis
The study of rapid reactions
Rapid mixing
The traditional experimental methods described above all assume that we can follow the reaction after its components have combined into a homogeneous mixture of known concentrations. But what can we do if the time required to complete the mixing process is comparable to or greater than the time needed for the reaction to run to completion? For reactions that take place in milliseconds, the standard approach since the 1950s has been to employ a flow technique of some kind. An early example was used to study fast gas-phase reactions in which one of the reactants is a free radical such as OH that can be produced by an intense microwave discharge acting on a suitable source gas mixture. This gas, along with the other reactant being investigated, is made to flow through a narrow tube at a known velocity.
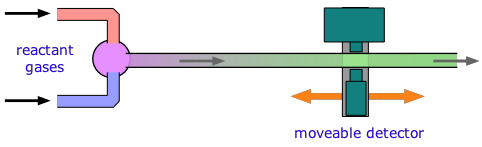
If the distance between the point at which the reaction is initiated and the product detector is known, then the time interval can be found from the flow rate. By varying this distance, the time required to obtain the maximum yield can then be determined.

Although this method is very simple in principle, it can be rather complicated in practice, as the illustration shows. Owing to the rather large volumes required, his method is more practical for the study of gas-phase reactions than for solutions, for which the stopped-flow method described below is generally preferred.
Stopped-flow and Quenched-flow methods
These are by far the most common means of studying fast solution-phase reactions over time intervals of down to a fraction of a millisecond. The use of reasonably simple devices is now practical even in student laboratory experiments. These techniques make it possible to follow not only changes in the concentrations of reactants and products, but also the buildup and decay of reaction intermediates.
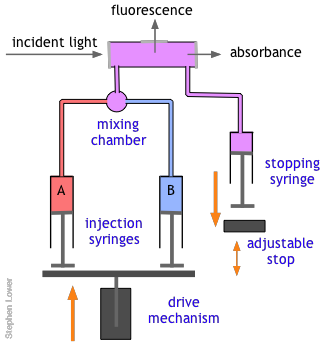

The basic stopped-flow apparatus consists of two or more coupled syringes that rapidly inject the reactants into a small mixing chamber and then through an observation cell that can be coupled to instruments that measure absorption, fluorescence, light scattering, or other optical or electrical properties of the solution. As the solution flows through the cell, it empties into a stopping syringe that, when filled, strikes a backstop that abruptly stops the flow. The volume that the stopping syringe can accept is adjusted so that the mixture in the cell has just become uniform and has reached a steady state; at this point, recording of the cell measurement begins and its change is followed.
Of course, there are many reactions that cannot be followed by changes in light absorption or other physical properties that are conveniently monitored. In such cases, it is often practical to quench (stop) the reaction after a desired interval by adding an appropriate quenching agent. For example, an enzyme-catalyzed reaction can be stopped by adding an acid, base, or salt solution that denatures (destroys the actvity of) the protein enzyme. Once the reaction has been stopped, the mixture is withdrawn and analyzed in an appropriate manner.
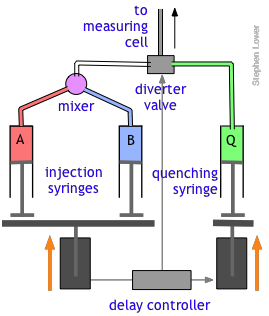
The quenched-flow technique works something like the stopped-flow method described above, with a slightly altered plumbing arrangement. The reactants A and B are mixed and fed directly through the diverter valve to the measuring cell, which is not shown in this diagram. After a set interval that can vary from a few milliseconds to 200 sec or more, the controller activates the quenching syringe and diverter valve, flooding the cell with the quenching solution.
Too fast to mix? No problem!
In order to investigate reactions that are complete in less than a millisecond, one can start with a pre-mixed sample in which one of active reactants is generated in situ. Alternatively, a rapid change in pressure or temperature can alter the composition of a reaction that has already achieved equilibrium.
Flash photolysis
Many reactions are known which do not take place in the absence of light whose wavelength is sufficiently short to supply the activation energy needed to break a bond, often leading to the creation of a highly reactive radical. A good example is the combination of gaseous Cl2 with H2, which procedes explosively when the system is illuminated with visible light. In flash photolysis, a short pulse of light is used to initiate a reaction whose progress can be observed by optical or other means.
Photolysis refers to the use of light to decompose a molecule into simpler units, often ions or free radicals. In contrast to thermolysis (decomposition induced by high temperature), photolysis is able to inject energy into a molecule almost instantaneously and can be much "cleaner", meaning that there are fewer side reactions that often lead to complex mixtures of products. Photolysis can also be highly specific; the wavelength of the light that triggers the reaction can often be adjusted to activate one particular kind of molecule without affecting others that might be present.
All this had been known for a very long time, but until the mid-1940's there was no practical way of studying the kinetics of the reactions involving the highly reactive species producd by photolysis. In 1945, Ronald Norrish of Cambridge University and his graduate student George Porter conceived the idea of using a short-duration flash lamp to generate gas-phase CH2 radicals, and then following the progress of the reaction of these radicals with other species by means of absorption spectroscopy.
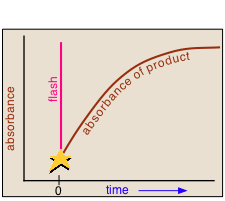
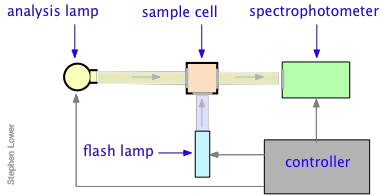
Norrish and Porter shared the 1967 Nobel Prize in Chemistry for this work.Nanosecond flash photolysis setup.
In a flash photolysis experiment, recording of the absorbance of the sample cell contents is timed to follow the flash by an interval that can be varied in order to capture the effects produced by the product or intermediate as it is formed or decays. Flash durations of around 1 millisecond permitted one to follow processes having lifetimes in the microsecond range, but the advent of fast lasers gradually extended this to picoseconds and femtoseconds.
Flash photolysis revolutionized the study of organic photochemistry, especially that relating to the chemistry of free radicals and other reactive species that cannot be isolated or stored, but which can easily be produced by photolysis of a suitable precursor. It has proven invaluable for understanding the complicated kinetics relating to atmospheric chemistry and smog formation. More recently, flash photolysis has become an important tool in biochemistry and cellular physiology.
Perturbation-relaxation methods
Many reactions, especially those that take place in solution, occur too rapidly to follow by flow techniques, and can therefore only be observed when they are already at equilibrium. The classical examples of such reactions are two of the fastest ones ever observed, the dissociation of water
\[2 H_2O → H_3O^+ + OH^–\]
and the formation of the triiodide ion in aqueous solution
\[I^– + I_2 → I_3^–\]
Reactions of these kinds could not be studied until the mid-1950s when techniques were developed to shift the equilibrium by imposing an abrupt physical change on the system.
For example, if the reaction A B is endothermic, then according to the Le Chatelier principle, subjecting the system to a rapid jump in temperature will shift the equilibrium state to one in which the product B has a higher concentration. The composition of the system will than begin to shift toward the new equilibrium composition at a rate determined by the kinetics of the process.
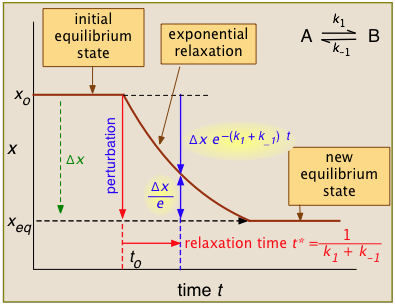
For the general case illustrated here, the quantity "x" being plotted is a measurable quantity such as light absorption or electrical conductivity that varies linearly with the composition of the system. In a first-order process, x will vary with time according to
xt = xo e–kt
After the abrupt perturbation at time to, the relaxation time t* is defined as the half-time for the return to equilibrium — that is, as the time required for xo to decrease by Δx/e = Δx/2.718. The derivation of t* and the relations highlighted in yellow can be found in most standard kinetics textbooks. Temperature jumps are probably most commonly used. They can be brought about in various ways:
This is the method that Manfred Eigen pioneered when, in the early 1960's, he measured the rate constant of what was then the fastest reaction ever observed:
H+ + OH– → H2O k = 1.3 × 1011 M–1 sec–1
A detailed description of the kinetics of this process can be seen here. Eigen shared the 1967 Nobel Prize in Chemistry with Porter and Norrish, who developed flash photolysis. Eigen's Nobel lecture, in keeping with his legendary sense of humor, was titled "immeasurably fast reactions". His later work has focused on self-organizing systems and the origin of life, molecular genetics, and neurology.
- high-voltage electric discharge: A capacitor, charged to 5-10 kV, is discharged through a solution to which an electrolyte has been added to provide a conductive path.
- laser irradiation: The sample is irradiated with a laser whose wavelength corresponds to an absorption peak in the sample. Infrared lasers are often used for this purpose.
- mixing of two pre-equilibrated solutions: Two solutions, otherwise identical but at different temperatures, are rapidly mixed in a stopped-flow type of apparatus. Although this method is not as fast, it has the advantage of allowing both negative and positive T-jumps. The device shown here uses 0.1-mL samples and provides jumps of up to ±40 C° over a few microseconds. Observation times, however, are limited to 1-2 milliseconds owing to thermal dissipation.
Pressure jumps
According to the Le Chatelier principle, a change in the applied pressure will shift the equilibrium state of any reaction which involves a change in the volume of a system. Aside from the obvious examples associated with changes in the number of moles of gases, there are many more subtle cases involving formation of complexes, hydration shells and surface adsorption, and phase changes. One area of considerable interest is the study of protein folding, which has implications in diseases such as Parkinson's and Alzheimer's.
The pressure-jump is applied to the cell through a flexible membrane that is activated by a high-pressure gas supply, or through an electrically-actuated piezoelectric crystal. The latter method is employed in the device shown here, which can produce P-jumps of around 1GPa over sub-millisecond time intervals.
More on P-jumps: this 1999 review covers many non-biochemical applications. See also this article on protein folding and aggregation.
Shock tubes: extreme jumping
When a change in pressure propagates through a gas at a rate greater than the ordinary compressions and rarefactions associated with the travel of sound, a moving front (a shock wave) of very high pressure forms. This in turn generates an almost instantaneous rise in the temperature that can approach several thousand degrees in magnitude.
A shock tube is an apparatus in which shock waves can be generated and used to study the kinetics of gas-phase reactions that are otherwise inaccessible to kinetic measurements. Since all molecules tend to dissociate at high temperatures, shock tubes are widely used to study dissociation processes and the chemistry of the resulting fragments. For example, the shock-induced decomposition of carbon suboxide provides an efficient means of investigating carbon atom reactions:
\[C_3O_2 → C + CO\]
Shock tube techniques are also useful for studying combustion reactions, including those that proceed explosively.
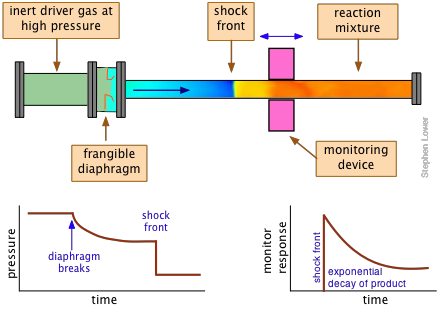
The shock tube itself consists of two sections separated by a breakable diaphragm of metal or plastic. One section is filled with a "driver" gas at a very high pressure, commonly helium, but often mixed with other inert gases to adjust the properties of the shock. The other, longer section of the tube contains the "driven" gas — the reactants — at a low pressure, usually less than 1 atmosphere.
The reaction is initiated by causing the diaphragm to rupture, either by means of a mechanical plunger or by raising the pressure beyond its bursting point. The kinetics of the reaction are monitored by means of an absorption or other optical monitoring device that is positioned at a location along the reaction tube that is appropriate to the time course of the reaction.