
24.7 Reactions of Amines
- Page ID
- 91035
Objectives
After completing this section, you should be able to
- write an equation to represent the reaction that takes place between ammonia, a primary or secondary amine, and an acid chloride.
- identify the product formed when a given amine reacts with a given acid chloride.
- identify the amine, the acid chloride, or both, needed to synthesize a given amide.
- write a reaction sequence to illustrate the overall conversion of an amine to an alkene via a Hofmann elimination.
- identify the alkene most likely to be formed when a given quaternary ammonium salt is heated with moist silver oxide (or silver hydroxide).
- deduce the structure of an unknown quaternary ammonium salt, given the identity of the alkene or alkenes produced when the salt is heated with moist silver oxide.
Make certain that you can define, and use in context, the key term below.
- Hofmann elimination
The alkylation and acylation of amines have been dealt with in previous sections: alkylation in Section 24.6 and acylation in Sections 21.4 and 21.5. Review these sections if necessary.
One explanation is given for Hofmann elimination further on in this reading. Another argument, based on the consideration of the structures of the possible transition states, has been suggested by Joseph Bunnett (Professor Emeritus, University of California, Santa Cruz) and is widely accepted. We can begin to understand Bunnett’s reasoning by considering the elimination products formed from by the dehydrohalogenation of a number of substituted hexanes in the reaction
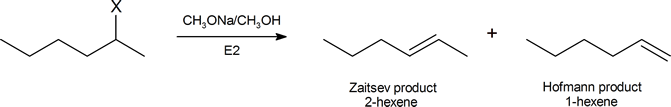
The table below describes the proportion of each product you would expect when X in the above reaction represents a given halogen.
| X | % 2‑hexene | % hexene |
|---|---|---|
| I | 81 | 19 |
| Br | 72 | 28 |
| Cl | 67 | 33 |
| F | 30 | 70 |
As we descend this table, the electron‑withdrawing ability of X increases, and the percentage of Hofmann product also increases. Given that the (CH3)3N+ group is strongly electron‑withdrawing, it is quite consistent for a compound of the type

to give 96% Hofmann product and 4% Zaitsev product in an E2 elimination reaction. But why does the proportion of Hofmann product increase as the electron‑withdrawing ability of X increases? Bunnett suggests that the transition state in an E2 process can vary from being “carbocation‑like” on one hand to “carbanion‑like” on the other.
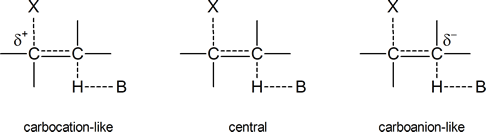
Figure24.1 Various transition states for an E2 elimination reaction
In a carbocation‑like transition state, the C$\ce{-}$X bond is broken to a greater extent than the C$\ce{-}$H bond; in the central transition state both, the C$\ce{-}$X bond and the C$\ce{-}$H bond are broken to an equal extent; and in the carbanion‑like transition state, the C$\ce{-}$H bond is broken to a greater extent than the C$\ce{-}$X bond. In the latter case, a partial negative charge will develop on the carbon atom to which the hydrogen is attached, hence the term “carbanion‑like.” As we already know, any species or transition state bearing a full or partial negative charge is stabilized by the presence of electron‑withdrawing groups and destabilized by the presence of electron‑releasing groups.
Let us now consider the two possible transition states for an E2 elimination of (CH3)3NH+ from a quaternary ammonium hydroxide,

One transition state leads to Zaitsev elimination, the other to Hofmann elimination.
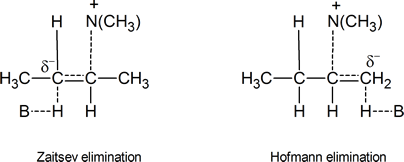
Figure24.2 Possible carbanion‑like transition states
In the transition state leading to the Zaitsev product, the carbon atom carrying the partial negative charge is bonded to an electron‑releasing methyl group. This is clearly a less favourable situation than the one that exists in the transition state leading to Hofmann elimination, where there are no electron‑releasing groups attached to the carbon atom bearing the partial negative charge. Bunnett’s argument is that carbanion‑like transition states are much more likely when the atom or group X in a compound such as
is strongly electron withdrawing, because the C$\ce{-}$X bond becomes stronger as the electron‑withdrawing ability of X increases.
Thus, as we go along the series X = I, Br, Cl, F, (CH3)3N+, the electron‑withdrawing ability of X increases, the C$\ce{-}$X bond becomes more difficult to break, the transition state becomes more carbanion‑like in character, and Hofmann elimination becomes more pronounced.
Alkylation
It is instructive to examine these nitrogen substitution reactions, using the common alkyl halide class of electrophiles. Thus, reaction of a primary alkyl bromide with a large excess of ammonia yields the corresponding 1º-amine, presumably by an SN2 mechanism. The hydrogen bromide produced in the reaction combines with some of the excess ammonia, giving ammonium bromide as a by-product. Water does not normally react with 1º-alkyl halides to give alcohols, so the enhanced nucleophilicity of nitrogen relative to oxygen is clearly demonstrated.
2 RCH2Br + NH3 (large excess)  RCH2NH2 + NH4(+) Br(–) RCH2NH2 + NH4(+) Br(–) |
It follows that simple amines should also be more nucleophilic than their alcohol or ether equivalents. If, for example, we wish to carry out an SN2 reaction of an alcohol with an alkyl halide to produce an ether (the Williamson synthesis), it is necessary to convert the weakly nucleophilic alcohol to its more nucleophilic conjugate base for the reaction to occur. In contrast, amines react with alkyl halides directly to give N-alkylated products. Since this reaction produces HBr as a co-product, hydrobromide salts of the alkylated amine or unreacted starting amine (in equilibrium) will also be formed.
2 RNH2 + C2H5Br RNHC2H5 + RNH3(+) Br(–)  RNH2C2H5(+) Br(–) + RNH2 RNH2C2H5(+) Br(–) + RNH2 |
Unfortunately, the direct alkylation of 1º or 2º-amines to give a more substituted product does not proceed cleanly. If a 1:1 ratio of amine to alkyl halide is used, only 50% of the amine will react because the remaining amine will be tied up as an ammonium halide salt (remember that one equivalent of the strong acid HX is produced). If a 2:1 ratio of amine to alkylating agent is used, as in the above equation, the HX issue is solved, but another problem arises. Both the starting amine and the product amine are nucleophiles. Consequently, once the reaction has started, the product amine competes with the starting material in the later stages of alkylation, and some higher alkylated products are also formed. Even 3º-amines may be alkylated to form quaternary (4º) ammonium salts. When tetraalkyl ammonium salts are desired, as shown in the following example, Hünig's base may be used to scavenge the HI produced in the three SN2 reactions. Steric hindrance prevents this 3º-amine (Hünig's base) from being methylated.
C6H5NH2 + 3 CH3I + Hünig's base C6H5N(CH3)3(+) I(–) + HI salt of Hünig's base
Acylation
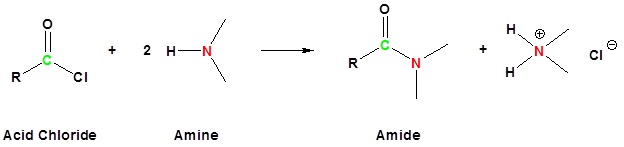
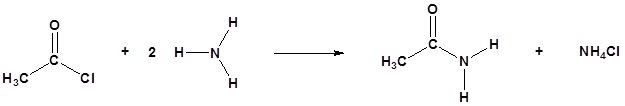
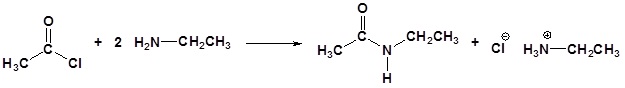
Amine functions seldom serve as leaving groups in nucleophilic substitution or base-catalyzed elimination reactions. Indeed, they are even less effective in this role than are hydroxyl and alkoxyl groups. In the case of alcohols and ethers, a useful technique for enhancing the reactivity of the oxygen function was to modify the leaving group (OH(–) or OR(–)) to improve its stability as an anion (or equivalent). This stability is conveniently estimated from the strength of the corresponding conjugate acids.
As noted earlier, 1º and 2º-amines are much weaker acids than alcohols, so it is not surprising that it is difficult to force the nitrogen function to assume the role of a nucleophilic leaving group. For example, heating an amine with HBr or HI does not normally convert it to the corresponding alkyl halide, as in the case of alcohols and ethers. In this context we note that the acidity of the putative ammonium leaving group is at least ten powers of ten less than that of an analogous oxonium species. The loss of nitrogen from diazonium intermediates is a notable exception in this comparison, due to the extreme stability of this leaving group (the conjugate acid of N2 would be an extraordinarily strong acid).
One group of amine derivatives that have proven useful in SN2 and E2 reactions is that composed of the tetraalkyl (4º-) ammonium salts. Most applications involving this class of compounds are eliminations, but a few examples of SN2 substitution have been reported.
C6H5–N(CH3)3(+) Br(–) + R-S(–) Na(+) |
acetone & heat |
R-S-CH3 + C6H5–N(CH3)2 + NaBr |
(CH3)4N(+) OH(–) |
heat |
CH3–OH + (CH3)3N |
Hofmann Elimination
Elimination reactions of 4º-ammonium salts are termed Hofmann eliminations. Since the counter anion in most 4º-ammonium salts is halide, this is often replaced by the more basic hydroxide ion through reaction with silver hydroxide (or silver oxide). The resulting hydroxide salt must then be heated (100 - 200 ºC) to effect the E2-like elimination of a 3º-amine. Example #1 below shows a typical Hofmann elimination. Obviously, for an elimination to occur one of the alkyl substituents on nitrogen must have one or more beta-hydrogens, as noted earlier in examining elimination reactions of alkyl halides.
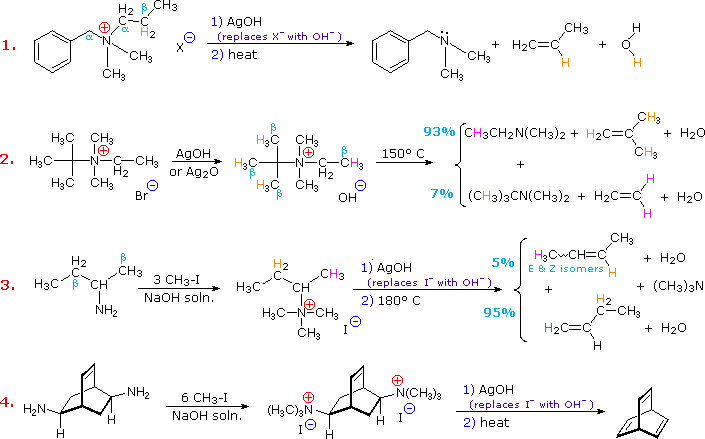
In example #2 above, two of the alkyl substituents on nitrogen have beta-hydrogens, all of which are on methyl groups (colored orange & magenta). The chief product from the elimination is the alkene having the more highly substituted double bond, reflecting not only the 3:1 numerical advantage of those beta-hydrogens, but also the greater stability of the double bond.
Example #3 illustrates two important features of the Hofmann elimination:
- Simple amines are easily converted to the necessary 4º-ammonium salts by exhaustive alkylation, usually with methyl iodide (methyl has no beta-hydrogens and cannot compete in the elimination reaction). Exhaustive methylation is shown again in example #4.
- When a given alkyl group has two different sets of beta-hydrogens available to the elimination process (colored orange & magenta here), the major product is often the alkene isomer having the less substituted double bond.
The tendency of Hofmann eliminations to give the less-substituted double bond isomer is commonly referred to as the Hofmann Rule, and contrasts strikingly with the Zaitsev Rule formulated for dehydrohalogenations and dehydrations. In cases where other activating groups, such as phenyl or carbonyl, are present, the Hofmann Rule may not apply. Thus, if 2-amino-1-phenylpropane is treated in the manner of example #3, the product consists largely of 1-phenylpropene (E & Z-isomers).
To understand why the base-induced elimination of 4º-ammonium salts behaves differently from that of alkyl halides it is necessary to reexamine the nature of the E2 transition state, first described for dehydrohalogenation. The energy diagram shown earlier for a single-step bimolecular E2 mechanism is repeated below.
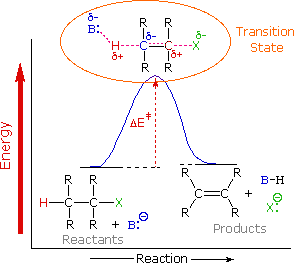
The E2 transition state is less well defined than is that of SN2 reactions. More bonds are being broken and formed, with the possibility of a continuum of states in which the extent of C–H and C–X bond-breaking and C=C bond-making varies. For example, if the bond to the leaving group (X) is substantially broken relative to the other bond changes, the transition state approaches that for an E1 reaction (initial ionization followed by a fast second step). At the other extreme, if the acidity of the beta-hydrogens is enhanced, then substantial breaking of C–H may occur before the other bonds begin to be affected. For most simple alkyl halides it was proper to envision a balanced transition state, in which there was a synchronous change in all the bonds. Such a model was consistent with the Zaitsev Rule.
When the leaving group X carries a positive charge, as do the 4º-ammonium compounds discussed here, the inductive influence of this charge will increase the acidity of both the alpha and the beta-hydrogens. Furthermore, the 4º-ammonium substituent is much larger than a halide or hydroxyl group and may perturb the conformations available to substituted beta-carbons. It seems that a combination of these factors acts to favor base attack at the least substituted (least hindered and most acidic) set of beta-hydrogens. The favored anti orientation of the leaving group and beta-hydrogen, noted for dehydrohalogenation, is found for many Hofmann eliminations; but syn-elimination is also common, possibly because the attraction of opposite charges orients the hydroxide base near the 4º-ammonium leaving group.
Three additional examples of the Hofmann elimination are shown in the following diagram. Example #1 is interesting in two respects. First, it generates a 4º-ammonium halide salt in a manner different from exhaustive methylation. Second, this salt is not converted to its hydroxide analog prior to elimination. A concentrated aqueous solution of the halide salt is simply dropped into a refluxing sodium hydroxide solution, and the volatile hydrocarbon product is isolated by distillation.
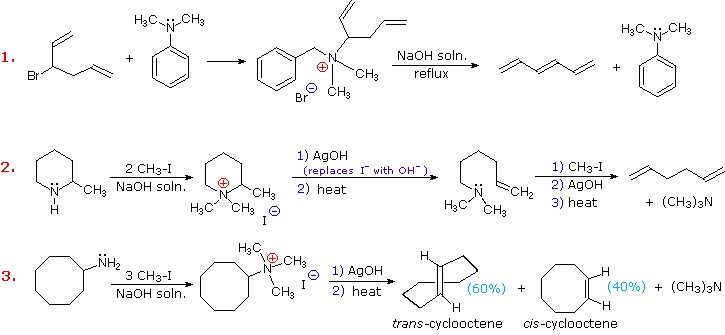
Example #2 illustrates an important aspect of the Hofmann elimination. If the nitrogen atom is part of a ring, then a single application of this elimination procedure does not remove the nitrogen as a separate 3º-amine product. In order to sever the nitrogen function from the molecule, a second Hofmann elimination must be carried out. Indeed, if the nitrogen atom was a member of two rings (fused or spiro), then three repetitions of the Hofmann elimination would be required to sever the nitrogen from the remaining molecular framework.
Example #3 is noteworthy because the less stable trans-cyclooctene is the chief product, accompanied by the cis-isomer. An anti-E2-transition state would necessarily give the cis-cycloalkene, so the trans-isomer must be generated by a syn-elimination. The cis-cyclooctene produced in this reaction could also be formed by a syn-elimination. Cyclooctane is a conformationally complex structure. Several puckered conformations that avoid angle strain are possible, and one of the most stable of these is shown on the right. Some eclipsed bonds occur in all these conformers, and transannular hydrogen crowding is unavoidable. Since the trimethylammonium substituent is large (about the size of tert-butyl) it will probably assume an equatorial-like orientation to avoid steric crowding. An anti-E2 transition state is likely to require an axial-like orientation of this bulky group, making this an unfavorable path.
Predict the preferred product of a Hofmann elimination with the following molecule.

- Answer
-
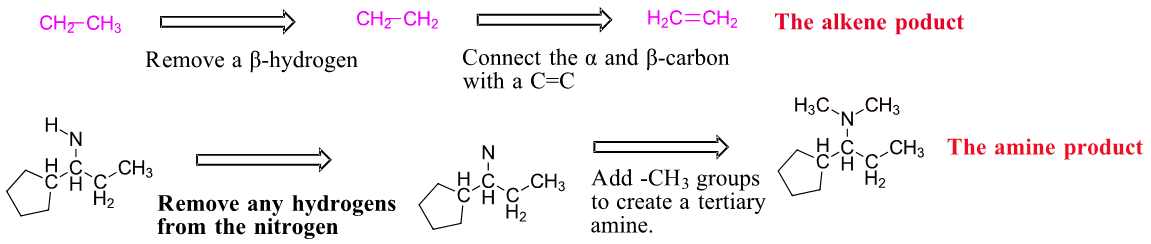
Predicting the product of a Hofmann elimination can be difficult so it is best done stepwise.
1) Find the least sterically hindered beta hydrogens. Beta hydrogens are on the second carbon away from the nitrogen. The order of preferred hydrogens are 1o > 2o > 3o. If the molecule is presented as a line structure it is usually beneficial to convert to a condensed structure first.

2) Break the C-N bond of the alkyl group which contains the preferred hydrogens. This will form two fragments.

3) Remove one of the original beta hydrogens in the alkyl fragment then draw a C=C between the alpha and beta carbon. This produces the alkene product of the Hofmann elimination. Sometimes the amine fragment of a Hofmann elimination is of interest. Remember that prior to elimination the amine is exhaustively alkylated with CH3I. This means any hydrogens attached to amine will starting material will be removed and the amine product will have enough methyl (-CH3) groups added to become a tertiary amine. In this case two methyl groups are added to the nitrogen. The end result are the two products of a Hofmann elimination.
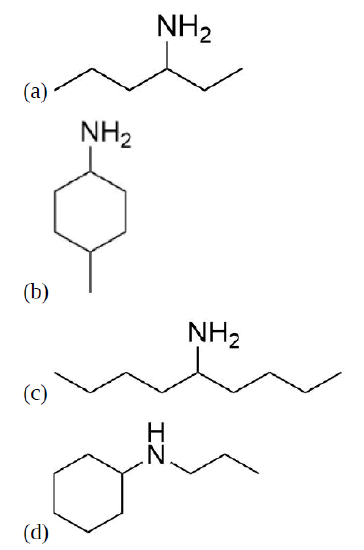
Exercises
Draw the product for a Hofmann elimination for each of the following molecules.
- Answer
-
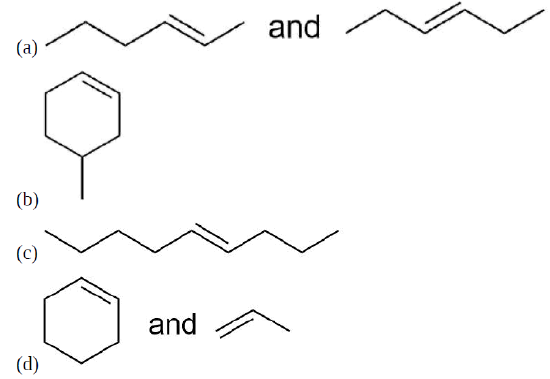
Draw the product of a Hofmann elimination for the following molecule.
- Answer
-
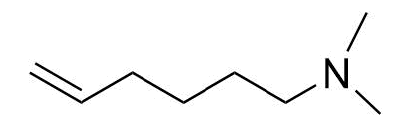
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry