
16.11 Rules of the Diels–Alder Reaction
- Page ID
- 28334

After completing this section, you should be able to
- determine whether or not a given compound would behave as a reactive dienophile in a Diels-Alder reaction.
- predict the stereochemistry of the product obtained from the reaction of a given diene with a given dienophile.
- recognize that in order to undergo a Diels-Alder reaction, a diene must be able to assume ans-cis geometry, and determine whether or not a given diene can assume this geometry.
Make certain that you can define, and use in context, the key terms below.
- dienophile
- dimerization
Make sure that you understand that the s-cis and s-trans forms of a diene such as 1,3-butadiene are conformers, not isomers. Note that some textbooks can confuse the issue further by referring to a compound such as (2Z, 4Z)-hexadiene as cis, cis-2,4-hexadiene, and saying that the most stable form of this compound is its s-trans conformer!
In fulfilling Objective 2, above, you must recognize that the Diels-Alder reaction is stereospecific.
Finally, note reaction B in the reading shows 1,3-cyclopentadiene reacting with another molecule of 1,3-cyclopentadiene. When the same compound acts as both diene and dienophile in a Diels-Alder reaction to couple it is a dimerization.
The Dienophile
In general, Diels-Alder reactions proceed fastest with electron-withdrawing groups on the dienophile (diene lover). Ethylene reacts slowly while propenal, ethyl propenoate, and other molecules shown below are highly reactive in a Diels-Alder reaction.
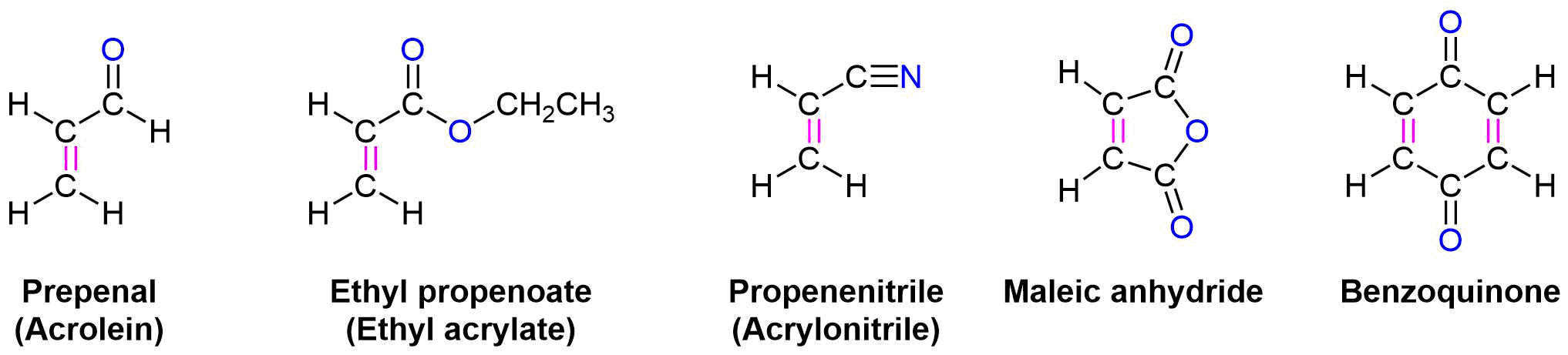
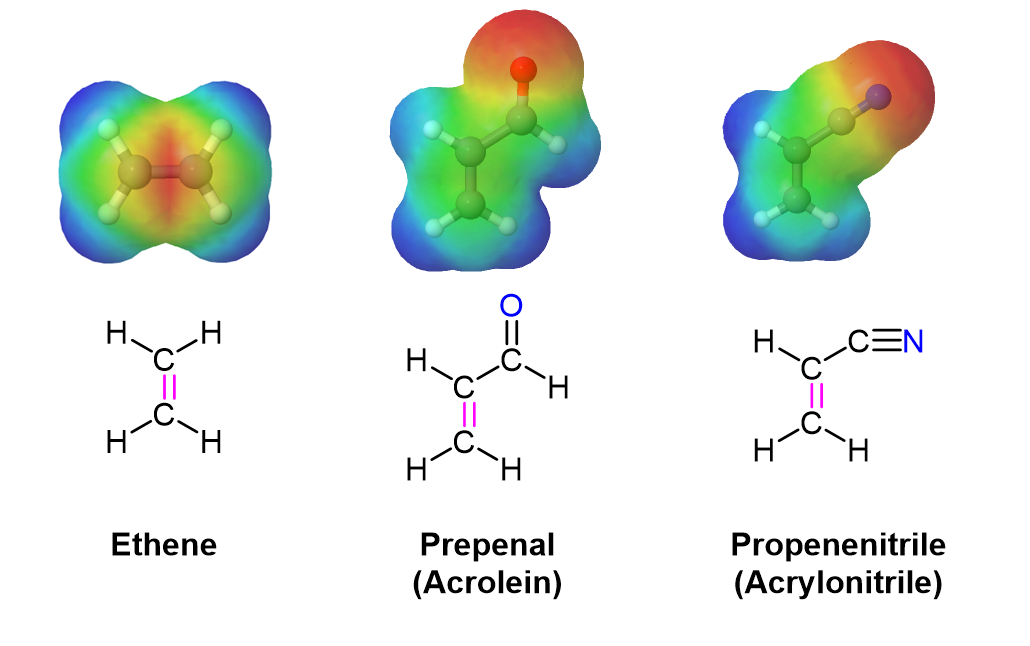
In much the same manner as electron-withdrawing substituents on a benzene ring, these are typically a double or triple bond in conjugation with the double bond in the dienophile. A resonance form can be drawn which places a positive charge in the dienophile double bond. This results in the double bond being less electron rich (greater electron density shown in Red/Orange) than ethylene. Electrostatic potential maps show that the electron-withdrawing groups pull electron density away from the double bond.
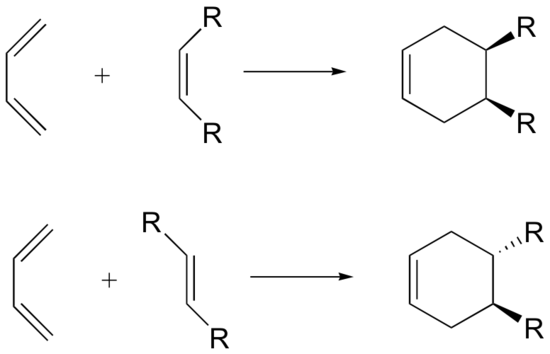
Stereochemistry of Diels-Alder (dienophile)
The Diels-Alder reaction is enormously useful for synthetic organic chemists, not only because ring-forming reactions are useful in general but also because in many cases multiple new stereocenters are formed, and the reaction is inherently stereospecific. During a Diels-Alder reaction the stereochemistry of the dienophile is retained in the product. A cis dienophile will generate a cyclohexene ring with cis (syn) substitution on the two carbons from the dienophile. Likewise a trans dienophile will generate a cyclohexene ring with trans (anti) substitution on these two carbon.
Example \(\PageIndex{1}\)
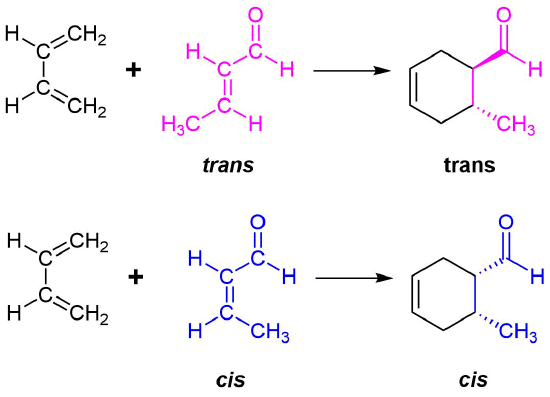
The retention of stereochemistry is due to the planar nature of both reactants and that the forming process is suprafacial (i.e. to or from the same face of each plane). This stereospecificity also confirms the concerted nature of the Diels-Alder mechanism. The drawing below illustrates this fact for the reaction of 1,3-butadiene with (E)-dicyanoethene. The trans relationship of the cyano groups in the dienophile is preserved in the six-membered ring of the adduct.
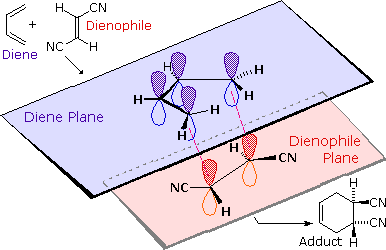
Another facet of the stereochemical retention of the dienophile is that only the endo product, rather than the alternative exo product, is formed. The words endo and exo are used to indicate relative stereochemistry when referring to bicyclic structures like substituted norbornanes. The endo position on a bicyclic structure refers to the position that is inside the concave shape of the larger (six-membered) ring. As you might predict, the exo position refers to the outside position. Diels-Alder reactions with cyclic dienes favor the formation of bicyclic structures in which substituents are in the endo position. Preference of the endo position is also a facet of the suprafacial nature of the Diels-Alder reaction. The orbital overlap required for the reaction is greater when the dienophile lies directly underneath the diene.

Alkynes can also serve as dienophiles in Diels-Alder reactions:

The Diene
In general, Diels-Alder reactions proceed fastest with electron-donating groups on the diene (eg. alkyl groups). The Diels-Alder reaction is a single step process, so the diene component must adopt an s-cis conformation in order for the end carbon atoms (#1 & #4) to bond simultaneously to the dienophile. For many acyclic dienes the s-trans conformer is more stable than the s-cis conformer (due to steric crowding of the end groups), but the two are generally in rapid equilibrium, permitting the use of all but the most hindered dienes as reactants in Diels-Alder reactions.
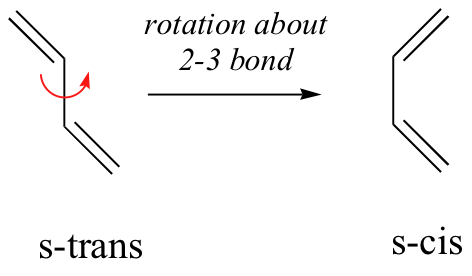
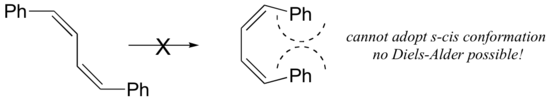
For some dienes, extreme steric hindrance causes the s-cis conformation to be highly strained, and for this reason such dienes do not readily undergo Diels-Alder reactions.
Cyclic dienes, on the other hand, are ‘locked’ in the s-cis conformation, and are especially reactive. The result of a Diels-Alder reaction involving a cyclic diene is a bicyclic structure:

Stereochemistry of Diels-Alder (diene)
The 1 and 4 Carbons in the diene have the possibility of forming two new stereocenters in the cyclohexene product. Similarly to the effects of dienophile stereochemistry, the positioning of substituents on the 1 and 4 carbons in the diene determine the stereochemistry in the product. The diene substituents can be thought of as being either cis (both facing in or both facing out) or trans and the stereochemistry is retained to form a cis or trans cyclohexene product.
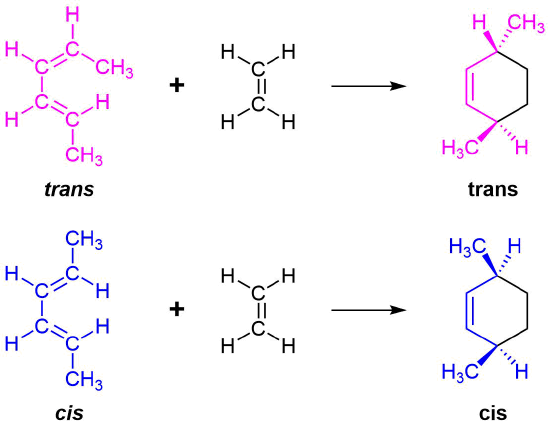
Examples \(\PageIndex{1}\)
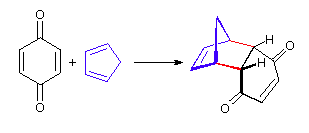
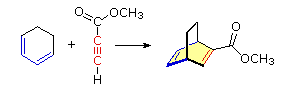
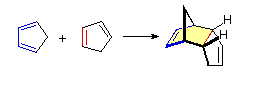
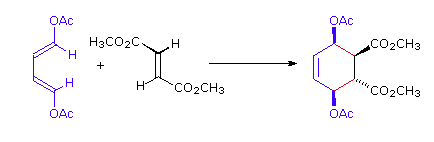
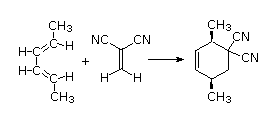
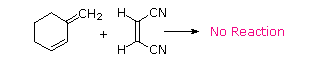
The Essential Characteristics of the Diels-Alder Cycloaddition Reaction:
- The reaction always creates a new six-membered ring.
- The diene component must be able to assume an s-cis conformation.
- Electron withdrawing groups on the dienophile facilitate reaction.
- Electron donating groups on the diene facilitate reaction.
- Steric hindrance at the bonding sites may inhibit or prevent reaction.
- The reaction is stereospecific with respect to substituent configuration in both the dienophile and the diene.
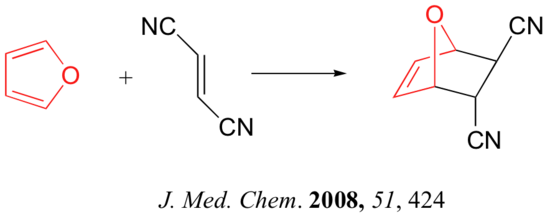
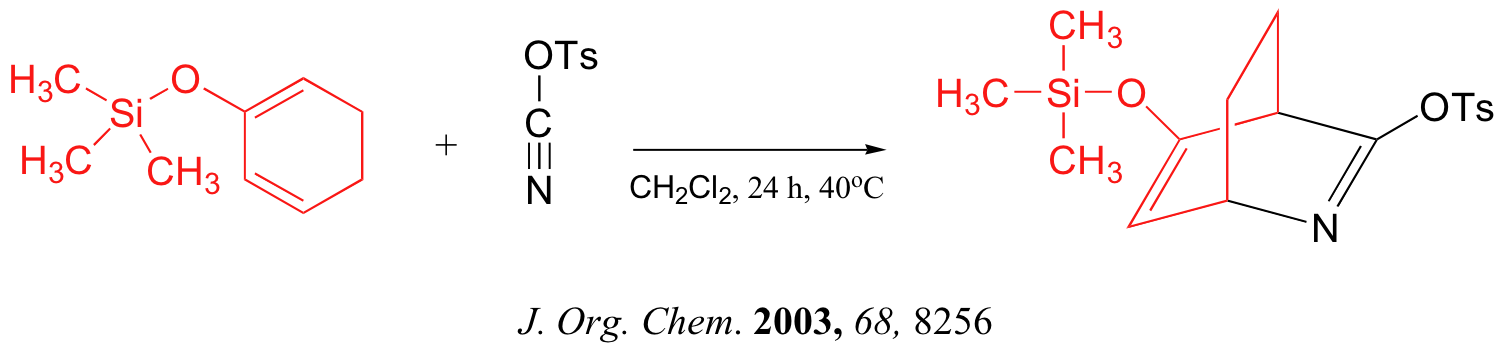
Predicting the Product of a Diels-Alder Reaction
Start by rotating the diene until it is in the s-cis conformation then point it towards the double bond of the dienophile. Remove the double bonds present in the diene and dieneophile. Connect carbons 1 and 4 of the the diene to a carbon in the dienophile double bond using a sigma bond to create a six-membered ring. Create a double bond between carbons 2 and 3 of diene.
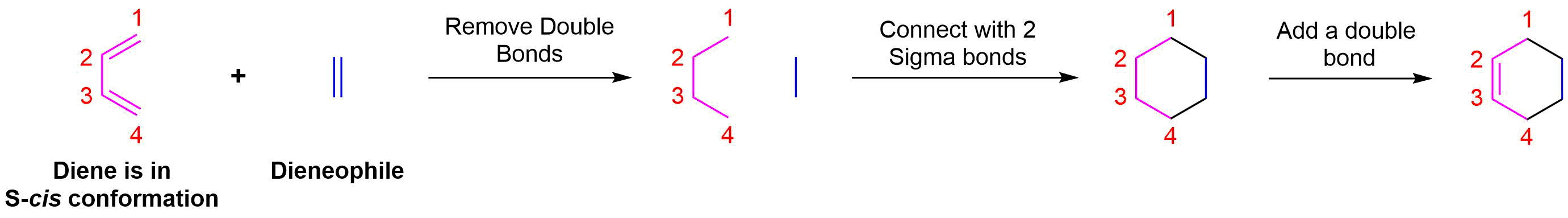
Determine if any substituents attached to either the double bond of the dieneophile or carbons 1 and 4 of the diene have a cis/trans conformation. If so make sure the substituents have the same configuration in the cycloalkene product.
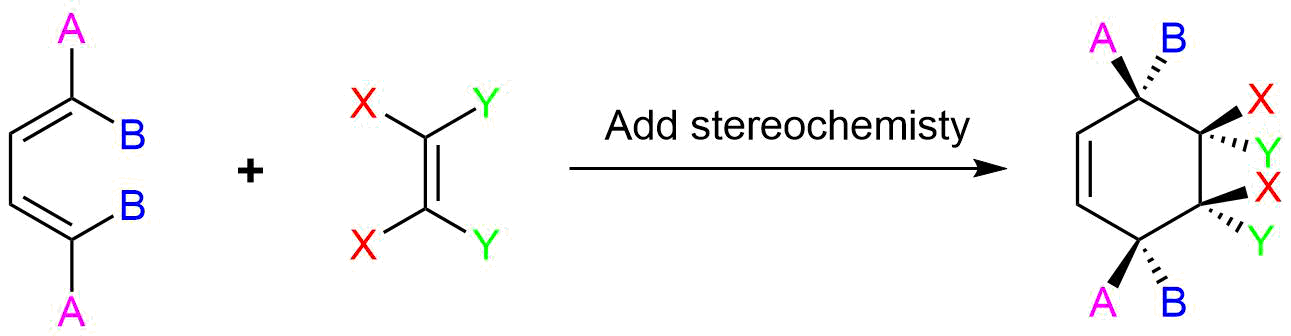
Predict the product of the following reaction:
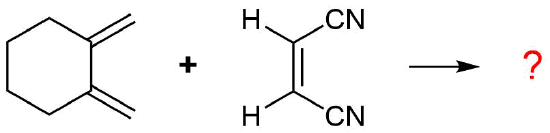
The diene is locked into an s-cis configuration which will promote the reaction. The ring portion of the diene will act as electron donating groups which will also promote the reaction. Because the diene already contained a ring the product will be bicyclic. The dienophile has two nitriles attached to it both of which are electron withdrawing. Since the two nitriles in the dieneophile are cis to each other the the two nitriles will be cis to each other in the product.
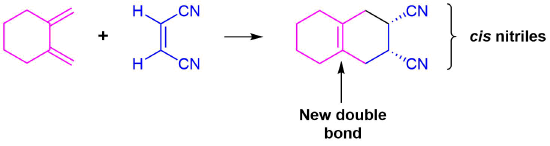
Predict the product of the following reaction:
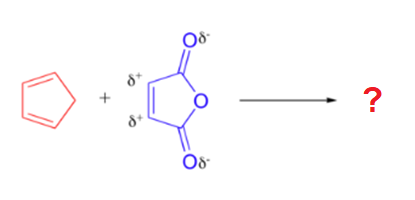
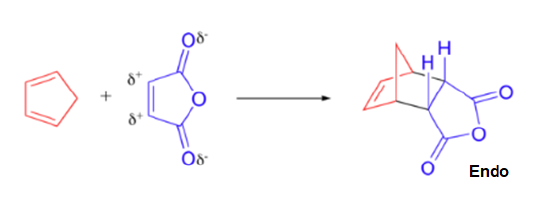
A particularly rapid Diels-Alder reaction takes place between cyclopentadiene and maleic anhydride. Cyclopentadiene is held in the required s-cis configuration so it will make a good diene for a Diels-Alder reaction. Maleic anhydride is also a very good dienophile, because the electron-withdrawing effect of the carbonyl groups causes the two alkene carbons to be electron-poor, and thus a good target for attack by the pi electrons in the diene. Since it is part of a ring, the double bond of maleic anhydride is in a cis configuration so the cyclohexene ring will also have a cis configuration. Lastly, the product will prefer the endo position.
Exercise \(\PageIndex{1}\)

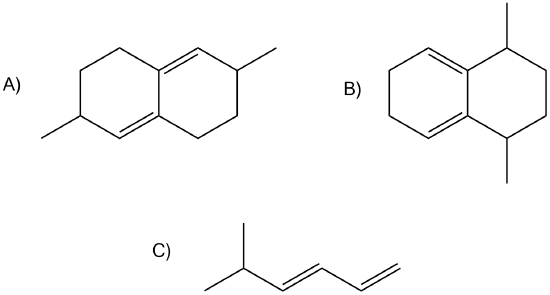
7) Of the following dienes, which are S-trans and which are s-cis? Of those that are s-trans, are they able to rotate to become s-cis?
8) Predict the product of the following reaction.
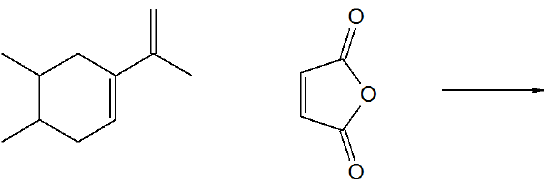
- Answer
-

7)
A) s-trans, unable to rotate to become s-cis
B) s-cis
C) s-trans, can rotate to become s-cis.
8)
.png?revision=1&size=bestfit&width=594&height=148)
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)
- Layne A. Morsch (University of Illinois Springfield)