
3.2: Conformations of Acyclic Hydrocarbons
- Page ID
- 319713
As we have mentioned previously, organic molecules exist in three dimensions. But the Lewis structures we draw are also not static – at room temperature (and even low temperature), molecules tumble, their bonds vibrate, and atoms rotate. Not all of these types of motion are the same in energy. Conformational analysis is a study of the energetics of rotational isomers, or conformers, that occur upon rotation of a single (\(σ\)) bond.
Conformations are different spatial arrangements of a molecule that are generated by rotation about single bonds. One molecule may have many different conformations. Some may be low in energy, others high. Some may be required for reactivity, while others may be completely inert. So, it is important to understand how conformations influence the properties of a molecule.
There are primarily two types of conformations we will be worried about here: a) conformations of acyclic hydrocarbons; and b) conformations of substituted hydrocarbons.
A. Conformations of acyclic hydrocarbons
Let’s consider ethane. One way of depicting ethane is by drawing all of the hydrogens and attaching some perspective, as in the Hawthorne structure (saw-horse) shown. If we look down the barrel of the C-C bond, what do we see?
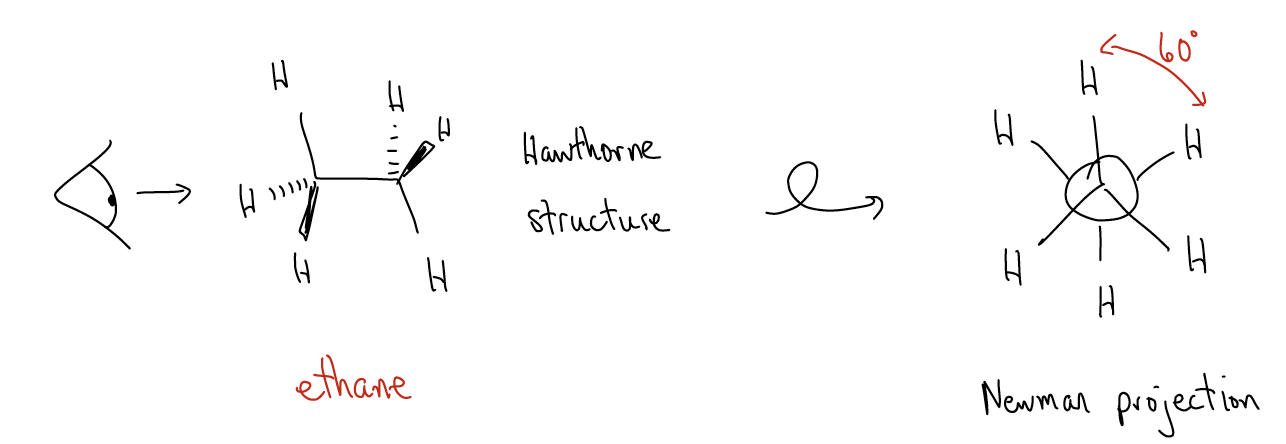
This drawing is known as a Newman projection. Is this a stable conformation? Why, yes it is. All of the H atoms are as far apart from each other as possible. The conformation in which the dihedral angle (HCCH) is 60° is called the staggered conformation.
Now, what if we were to rotate the rear carbon 60° clockwise without rotating the front carbon? What changes would we see? Draw the Newman projection:
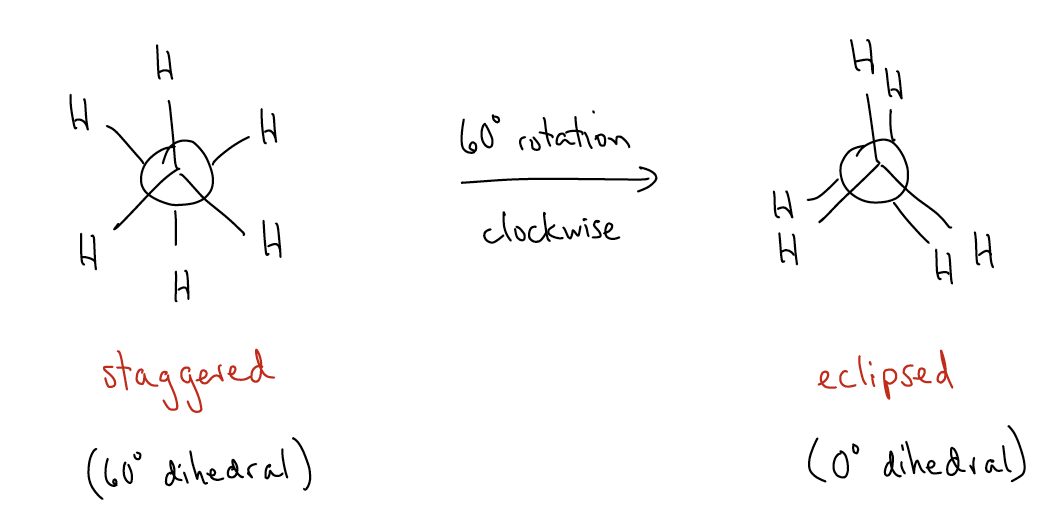
In this conformation, known as the eclipsed conformation, the dihedral angle is now 0°. This conformation is higher in energy than the staggered conformation by 2.9 kcal/mol. But, why is it higher? There are two reasons, both of which are electronic in nature:
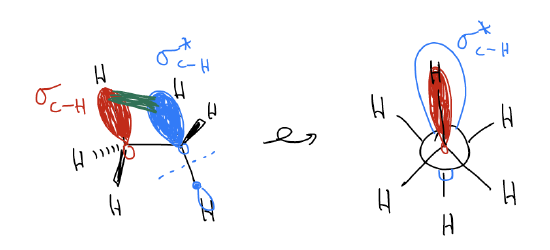
1. There is electron repulsion, known as torsional strain, between the overlapping electrons clouds of the two \(σ_{C-H}\) bonds. Like charges repel, so when they are close together, there is an increase in energy.
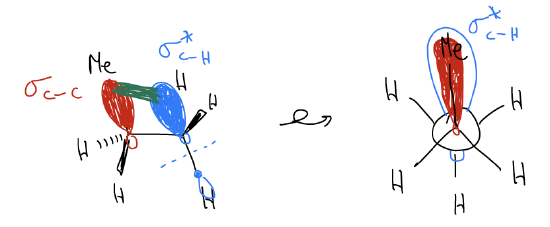
2. In the staggered conformation, electrons can delocalize from the \(σ_{C-H}\) into the \(σ^{*}_{C-H}\). This is known as hyperconjugation, or what I’m going to call \(σ\)-donation. This delocalization does not exist in the eclipsed conformation. Electrons from the \(σ_{C-H}\) are aligned correctly for overlap through space with the \(σ^{*}_{C-H}\), which leads to stabilization (think about it as electrons liking to be spread out more).
How would this look if we drew a potential energy diagram? As we’ve said, the difference in energy between the eclipsed and staggered conformations is 2.9 kcal/mol, which is the activation energy (Ea) required to proceed from one staggered conformation to the closest eclipsed conformation. Since there are three eclipsed conformations, we can say that each \(σ_{C-H}\)eclipsing interaction is approximately 0.9-1.0 kcal/mol. At room temperature, there is more than enough energy to overcome this barrier. The point of maximal potential energy, known as the transition state, is a transient species. It is fleeting and we cannot detect it, so eclipsed conformations will always funnel into the next low energy conformation.
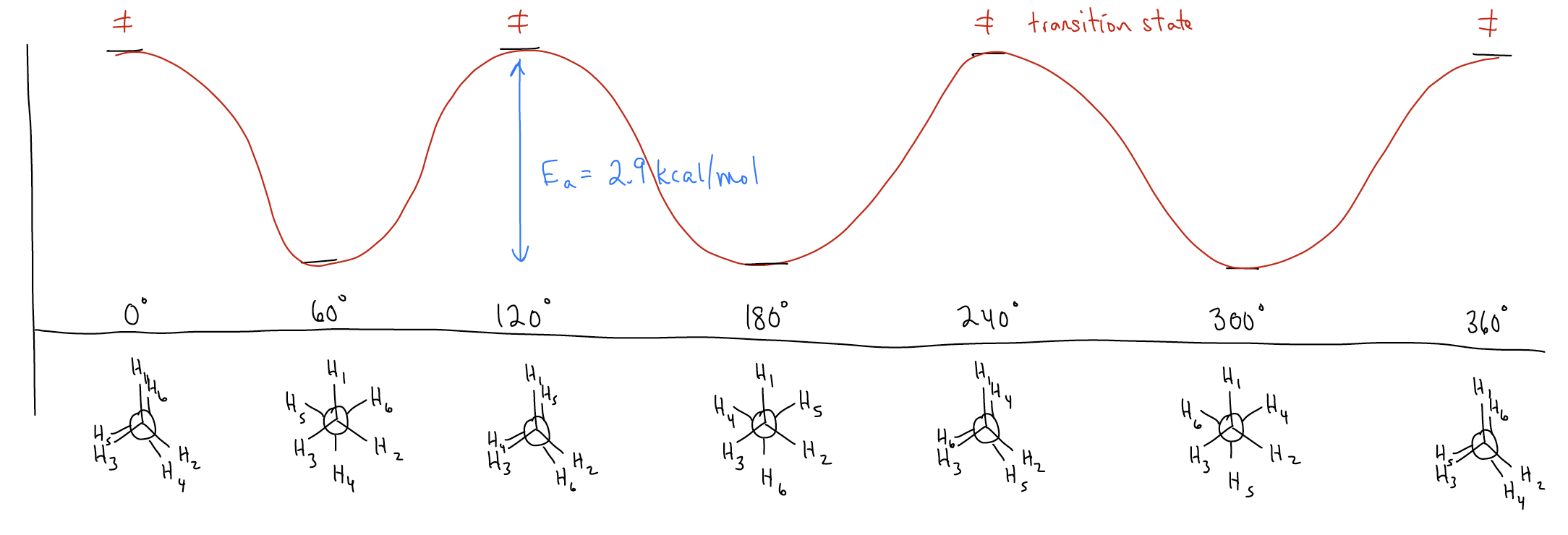
What does this energy different tell you about the relative distribution of conformations of ethane? In other words, what is the ratio between the staggered and eclipsed conformations? Recall:
ΔG = - RT ln K
1.9 kcal/mol = - (1.986 x 10-3 kcal/(K*mol)) x 298 K x ln K
K = 0.007446 therefore the ratio > 99:1
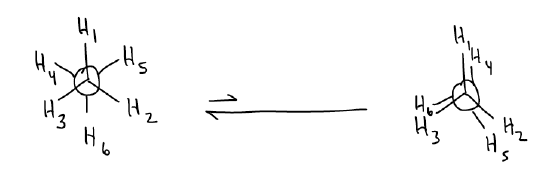
Let’s now add one more level of complexity. Draw the most stable conformation of propane and rotate around the C-C bond to form a potential energy diagram:
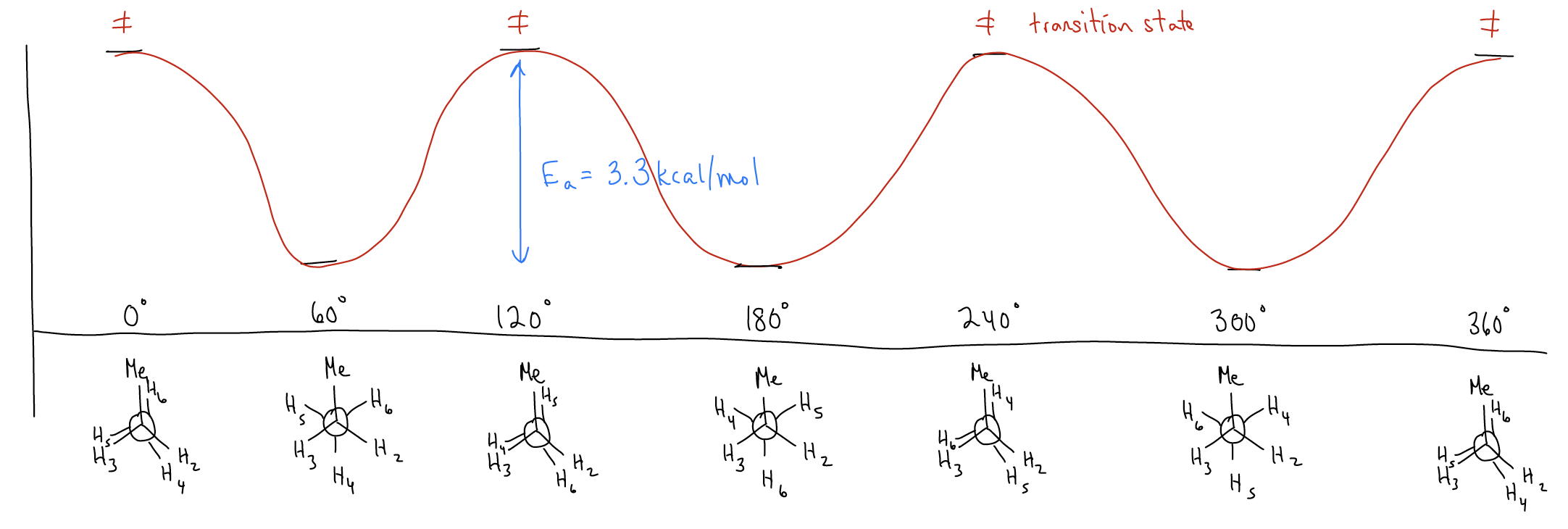
The activation energy is now 3.3 kcal/mol! Why is it higher than ethane? Two principles continue to influence the conformation:
1. Torsional strain – now, it is between \(σ_{C-C}\) (which is a little longer) and \(σ_{C-H}\).
2. Hyperconjugation (\(σ\)-donation) – now, the delocalization is between \(σ_{C-C}\) and \(σ^{*}_{C-H}\). Since a \(σ_{C-C}\) bond is a slightly better donor than \(σ_{C-H}\), there is a greater stabilization effect.
Because of these effects, the difference between the eclipsed and staggered conformers are maybe not as great as you would expect by adding a much bigger methyl group.
Let’s now go one atom longer. Draw the three most stable conformations for butane:
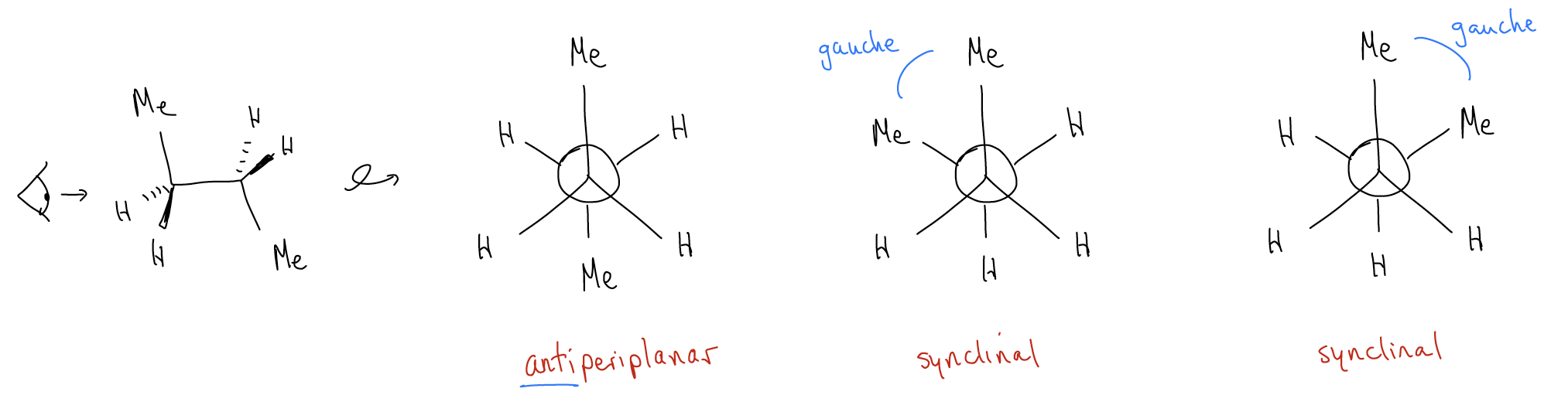
Did you notice that not all of the staggered conformations look the same? The conformation in which the dihedral angle between the methyl groups is 180° is called antiperiplanar, and we say that the methyl groups are anti. The conformation where the dihedral angle is 60° is called synclinal, and we say that the methyl groups are gauche. There are two synclinal conformations and one antiperiplanar conformation, all of which are staggered, and therefore low in energy. What would a potential energy diagram look like in this case?
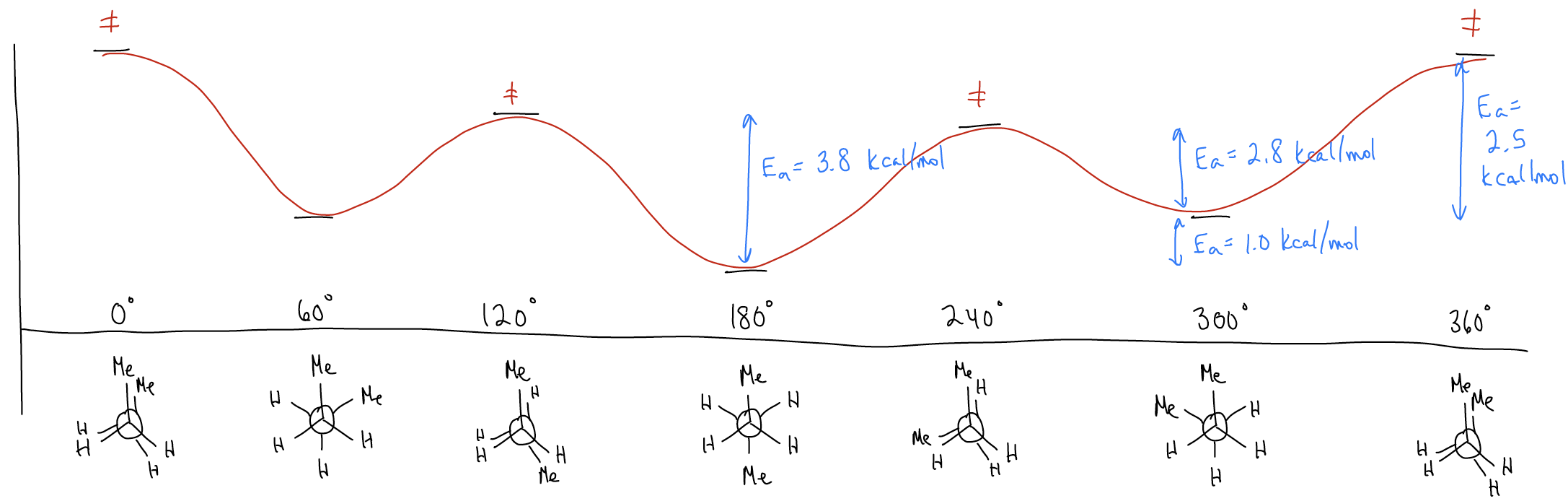
Notice that the energy differences between the various conformers are quite different this time. What accounts for this?
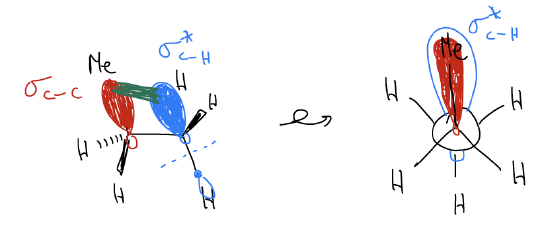
1. Torsional strain - now, it is between \(σ_{C-C}\) (which is a little longer) and both \(σ_{C-H}\) and \(σ_{C-C}\).
2. Hyperconjugation (\(σ\)-donation) – now, one particular delocalization is between \(σ_{C-C}\) and \(σ^{*}_{C-C}\). This is highly stabilizing since not only is a \(σ_{C-C}\) bond a slightly better donor than \(σ_{C-H}\), but because the \(σ^{*}_{C-C}\) is lower in energy than the \(σ^{*}_{C-H}\) (consider the effects of electronegativity and bond length). Add up all of the other stabilizing effects, and the energy of the staggered conformations go down.
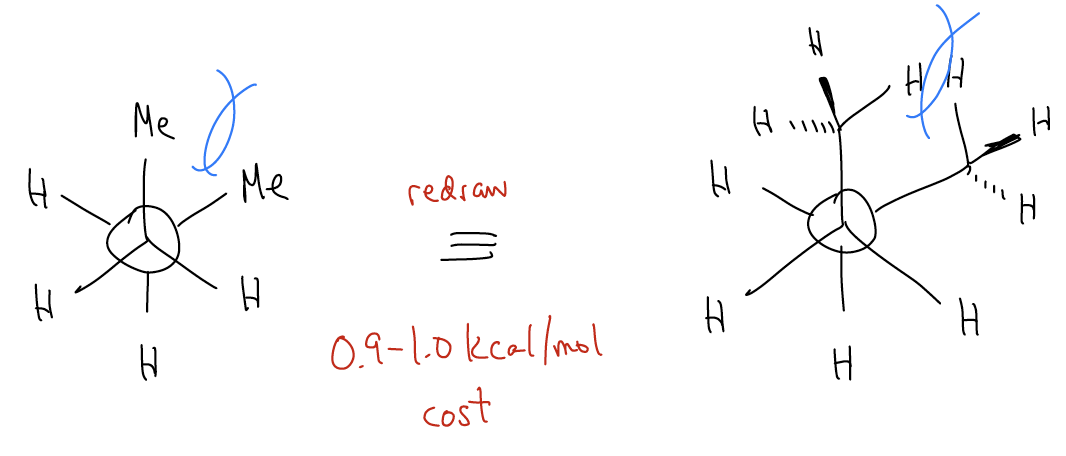
3. Steric hindrance - occurs when two groups are too close to each other. Their electron clouds overlap too much, creating electronic repulsion. This is different from torsional strain because the atoms themselves are bumping into each other, not just the bonds. This should explain why the antiperiplanar conformation is more stable (by about 0.9 kcal/mol) than the synclinal conformations. A gauche interaction, which is true steric strain, between methyl groups costs about 0.9 kcal/mol.
So, let’s summarize our findings:
H/H eclipsed interactions = 0.9-1.0 kcal/mol per bond
H/CH3 eclipsed interaction = 1.4 kcal/mol per bond
CH3/ CH3 eclipsed interaction = 2.4 kcal/mol per bond
CH3/ CH3 gauche interactions = 1.0 kcal/mol per bond
Look back at your potential energy diagrams to convince yourself that these values are accurate! What this all should mean is that in alkanes, the antiperiplanar staggered conformation will be the lowest in energy – this is why we draw alkanes in line-angle notation with a zig-zag! After all, the zig-zag is an antiperiplanar arrangement for the carbon skeleton.