
11.6: Synthesis of Alcohols - Review
- Page ID
- 182995
Alcohols are prepared by SN2 & SN1 (solvolysis) reactions
Alkyl halides can be converted to alcohols by using SN2 reactions with OH- as a nucleophile. Substrates that undergo substitution by SN1 reaction can be converted to alcohols using water as the nucleophile (and it can even be the solvent). Recall that SN1 reactions are promoted in polar, protic solvents.
Alcohols from alkenes
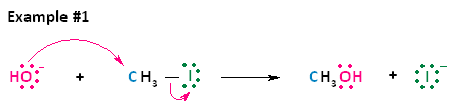
Oxymercuration is a special electrophilic addition. It is anti-stereospecific and regioselective. Regioselectivity is a process in which the substituents choses one direction it prefers to be attached to over all the other possible directions. The good thing about this reaction is that there are no carbocation rearrangement due to stabilization of the reactive intermediate. Similar stabilization is also seen in bromination addition to alkenes.
Carbocation rearrangement is a process in which the carbocation intermediate can form a more stable ion. With carbocation rearrangement, the reaction would not be able to hydrate quickly under mild conditions and be produced in high yields. This reaction is very fast and proceeds with 90% yield.
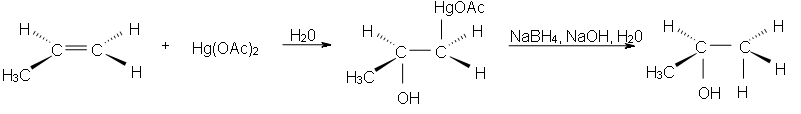
Hydroboration-Oxidation is a two step pathway used to produce alcohols. The reaction proceeds in an Anti-Markovnikov manner, where the hydrogen (from BH3 or BHR2) attaches to the more substituted carbon and the boron attaches to the least substituted carbon in the alkene bouble bond. Furthermore, the borane acts as a lewis acid by accepting two electrons in its empty p orbital from an alkene that is electron rich. This process allows boron to have an electron octet. A very interesting characteristic of this process is that it does not require any activation by a catalyst. The Hydroboration mechanism has the elements of both hydrogenation and electrophilic addition and it is a stereospecific (syn addition), meaning that the hydroboration takes place on the same face of the double bond, this leads cis stereochemistry.
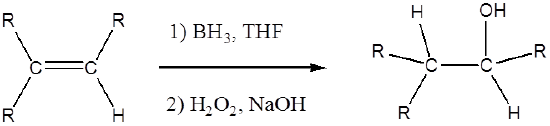
Diols from alkenes
Epoxides may be cleaved by aqueous acid to give glycols that are often diastereomeric with those prepared by the syn-hydroxylation reaction described above. Proton transfer from the acid catalyst generates the conjugate acid of the epoxide, which is attacked by nucleophiles such as water in the same way that the cyclic bromonium ion described above undergoes reaction. The result is anti-hydroxylation of the double bond, in contrast to the syn-stereoselectivity of the earlier method. In the following equation this procedure is illustrated for a cis-disubstituted epoxide, which, of course, could be prepared from the corresponding cis-alkene. This hydration of an epoxide does not change the oxidation state of any atoms or groups.
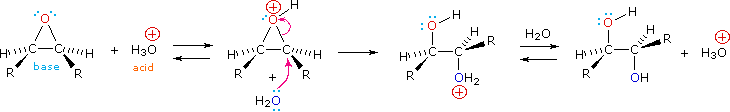
Osmium tetroxide oxidizes alkenes to give glycols through syn addition. A glycol, also known as a vicinal diol, is a compound with two -OH groups on adjacent carbons.

The reaction with \(OsO_4\) is a concerted process that has a cyclic intermediate and no rearrangements. Vicinal syn dihydroxylation complements the epoxide-hydrolysis sequence which constitutes an anti dihydroxylation of an alkene. When an alkene reacts with osmium tetroxide, stereocenters can form in the glycol product. Cis alkenes give meso products and trans alkenes give racemic mixtures.
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)