
6.11: The Hammond Postulate and Transition States
- Page ID
- 182910
Learning Objective
- explain and apply Hammond's Postulate to substitution reactions
Now, back to transition states. Chemists are often very interested in trying to learn about what the transition state for a given reaction looks like, but addressing this question requires an indirect approach because the transition state itself cannot be observed. In order to gain some insight into what a particular transition state looks like, chemists often invoke the Hammond postulate, which states that a transition state resembles the structure of the nearest stable species. For an exergonic reaction, therefore, the transition state resembles the reactants more than it does the products.
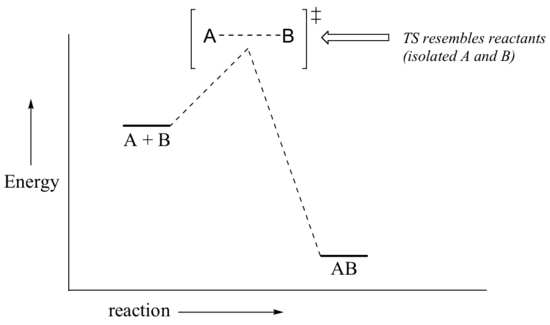
If we consider a hypothetical exergonic reaction between compounds A and B to form AB, the distance between A and B would be relatively large at the transition state, resembling the starting state where A and B are two isolated species. In the hypothetical endergonic reaction between C and D to form CD, however, the bond formation process would be much further along at the TS point, resembling the product.
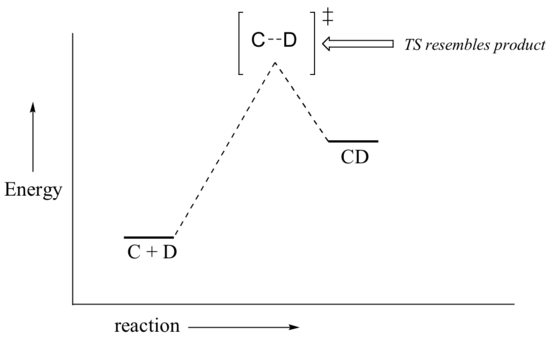
The Hammond Postulate is a very simplistic idea, which relies on an assumption that potential energy surfaces are parabolic. Although such an assumption is not rigorously true, it is fairly reliable and allows chemists to make energetic arguments about transition states by employing arguments about the stability of a related species. Since the formation of a reactive intermediate is very reliably endergonic, arguments about the stability of reactive intermediates can serve as proxy arguments about transition state stability.
The Hammond Postulate and the SN1 Reaction
The Hammond postulate suggests that the activation energy of the rate-determining first step will be inversely proportional to the stability of the carbocation intermediate. The stability of carbocations is shown qualitatively below:
Carbocation Stability
\[\ce{CH3(+) < CH3CH2(+) < (CH3)2CH(+) ≈ CH2=CH-CH2(+) < C6H5CH2(+) ≈ (CH3)3C(+)}\]
Consequently, we expect that 3º-alkyl halides will be more reactive than their 2º and 1º-counterparts in reactions that follow an SN1 mechanism. This is opposite to the reactivity order observed for the SN2 mechanism. Allylic and benzylic halides are exceptionally reactive by either mechanism.
Contributors
Prof. Steven Farmer (Sonoma State University)
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)