
3.12: Infrared Spectroscopy
- Page ID
- 201107
After completing this section, you should be able to
- identify (by wavelength, wavenumber, or both) the region of the electromagnetic spectrum which is used in infrared (IR) spectroscopy.
- interconvert between wavelength and wavenumber.
- discuss, in general terms, the effect that the absorption of infrared radiation can have on a molecule.
Make certain that you can define, and use in context, the key terms below.
- infrared spectrum
- wavenumber (reciprocal centimetres)
Notice that the scale at the bottom of the infrared spectrum for 2-hexanone shown is calibrated in wavenumbers (cm−1). A wavenumber is the reciprocal of a wavelength (1/λ); thus, a wavenumber of 1600 cm−1 corresponds to a wavelength of
Organic chemists find it more convenient to deal with wavenumbers rather than wavelengths when discussing infrared spectra.
You will obtain infrared spectra for a number of the compounds you will synthesize in the laboratory component of this course.
The inverted peaks observed in the spectra correspond to molecular stretching and bending vibrations that only occur at certain quantized frequencies. When infrared radiation matching these frequencies falls on the molecule, the molecule absorbs energy and becomes excited. Eventually the molecule returns to its original (ground) state, and the energy which was absorbed is released as heat.
Infrared Spectroscopy
The full range of electromagnetic radiation wavelengths is referred to as the electromagnetic spectrum.
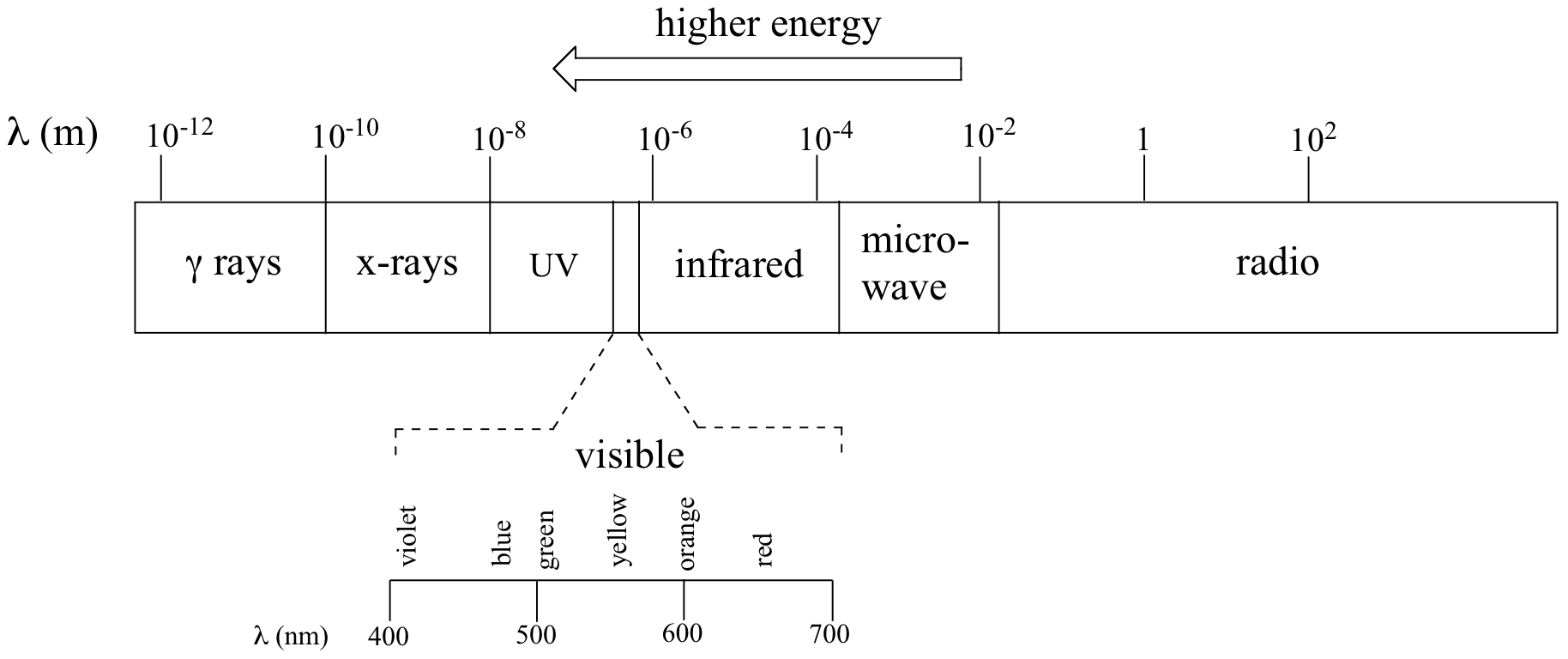
Notice in the figure above that infrared light is lower energy than visible light. The wavelengths of infrared radiation are between 0.8 and 250 μm. The units that are typically used for infrared spectroscopy are wavenumbers (which is cm-1). IR spectroscopy analyzes radiation between 40 to 13,000 cm-1. But what type of excitation is occurring when infrared radiation is absorbed by a molecule?
Covalent bonds in organic molecules are not rigid sticks – rather, they behave more like springs. At room temperature, organic molecules are always in motion, as their bonds stretch, bend, and twist. These complex vibrations can be broken down mathematically into individual vibrational modes, a few of which are illustrated below.
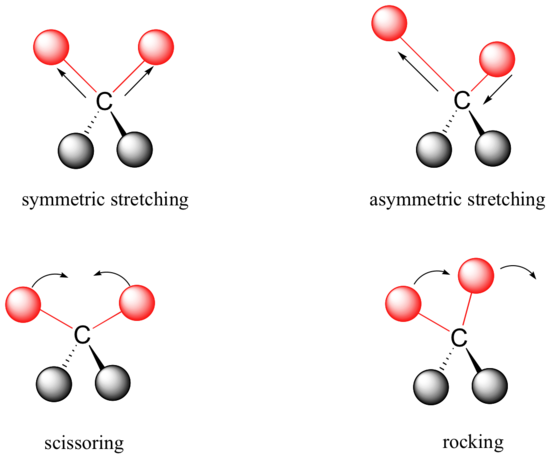
The energy of molecular vibration is quantized rather than continuous, meaning that a molecule can only stretch and bend at certain 'allowed' frequencies. If a molecule is exposed to electromagnetic radiation that matches the frequency of one of its vibrational modes, it will in most cases absorb energy from the radiation and jump to a higher vibrational energy state - what this means is that the amplitude of the vibration will increase, but the vibrational frequency will remain the same. The difference in energy between the two vibrational states is equal to the energy associated with the wavelength of radiation that was absorbed. It turns out that it is the infrared region of the electromagnetic spectrum which contains frequencies corresponding to the vibrational frequencies of organic bonds.
Let's take 2-hexanone as an example. Picture the carbonyl bond of the ketone group as a spring. This spring is constantly bouncing back and forth, stretching and compressing, pushing the carbon and oxygen atoms further apart and then pulling them together. This is the stretching mode of the carbonyl bond. In the space of one second, the spring 'bounces' back and forth 5.15 x 1013 times - in other words, the ground-state frequency of carbonyl stretching for a the ketone group is about 5.15 x 1013 Hz.
If our ketone sample is irradiated with infrared light, the carbonyl bond will specifically absorb light with this same frequency, which by equations 4.1 and 4.2 corresponds to a wavelength of 5.83 x 10-6 m and an energy of 4.91 kcal/mol. When the carbonyl bond absorbs this energy, it jumps up to an excited vibrational state.
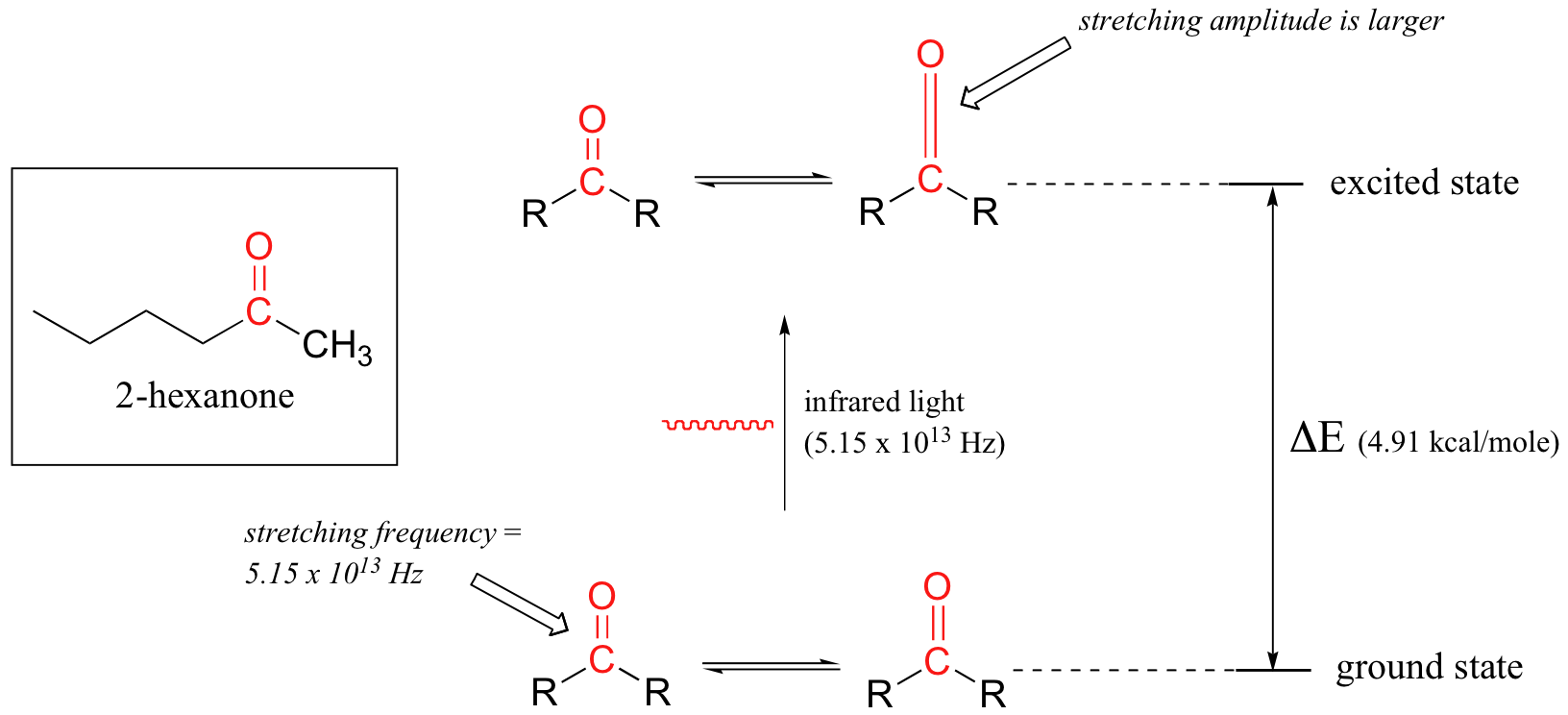
The value of ΔE - the energy difference between the low energy (ground) and high energy (excited) vibrational states - is equal to 4.91 kcal/mol, the same as the energy associated with the absorbed light frequency. The molecule does not remain in its excited vibrational state for very long, but quickly releases energy to the surrounding environment in form of heat, and returns to the ground state.
With an instrument called an infrared spectrophotometer, we can 'see' this vibrational transition. In the spectrophotometer, infrared light with frequencies ranging from about 1013 to 1014 Hz is passed though our sample of cyclohexane. Most frequencies pass right through the sample and are recorded by a detector on the other side.
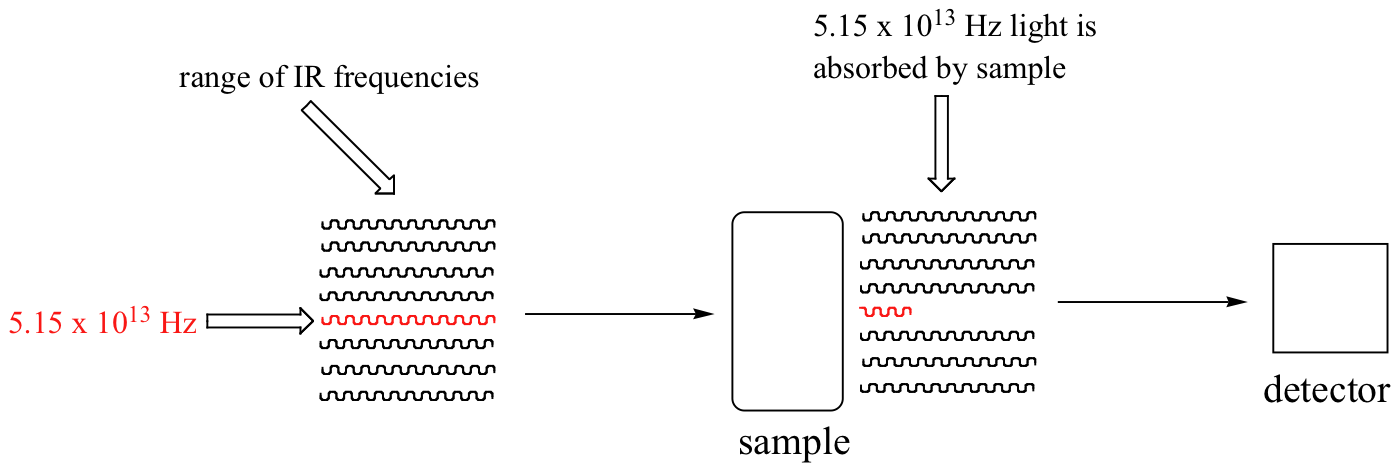
Our 5.15 x 1013 Hz carbonyl stretching frequency, however, is absorbed by the 2-hexanone sample, and so the detector records that the intensity of this frequency, after having passed through the sample, is something less than 100% of its initial intensity.
The vibrations of a 2-hexanone molecule are not, of course, limited to the simple stretching of the carbonyl bond. The various carbon-carbon bonds also stretch and bend, as do the carbon-hydrogen bonds, and all of these vibrational modes also absorb different frequencies of infrared light.
The power of infrared spectroscopy arises from the observation that different functional groups have different characteristic absorption frequencies. The carbonyl bond in a ketone, as we saw with our 2-hexanone example, typically absorbs in the range of 5.11 - 5.18 x 1013 Hz, depending on the molecule. The carbon-carbon triple bond of an alkyne, on the other hand, absorbs in the range 6.30 - 6.80 x 1013 Hz. The technique is therefore very useful as a means of identifying which functional groups are present in a molecule of interest. If we pass infrared light through an unknown sample and find that it absorbs in the carbonyl frequency range but not in the alkyne range, we can infer that the molecule contains a carbonyl group but not an alkyne.
Some bonds absorb infrared light more strongly than others, and some bonds do not absorb at all. In order for a vibrational mode to absorb infrared light, it must result in a periodic change in the dipole moment of the molecule. Such vibrations are said to be infrared active. In general, the greater the polarity of the bond, the stronger its IR absorption. The carbonyl bond is very polar, and absorbs very strongly. The carbon-carbon triple bond in most alkynes, in contrast, is much less polar, and thus a stretching vibration does not result in a large change in the overall dipole moment of the molecule. Alkyne groups absorb rather weakly compared to carbonyls.
Some kinds of vibrations are infrared inactive. The stretching vibrations of completely symmetrical double and triple bonds, for example, do not result in a change in dipole moment, and therefore do not result in any absorption of light (but other bonds and vibrational modes in these molecules do absorb IR light).
Now, let's look at some actual output from IR spectroscopy experiments. Below is the IR spectrum for 2-hexanone.
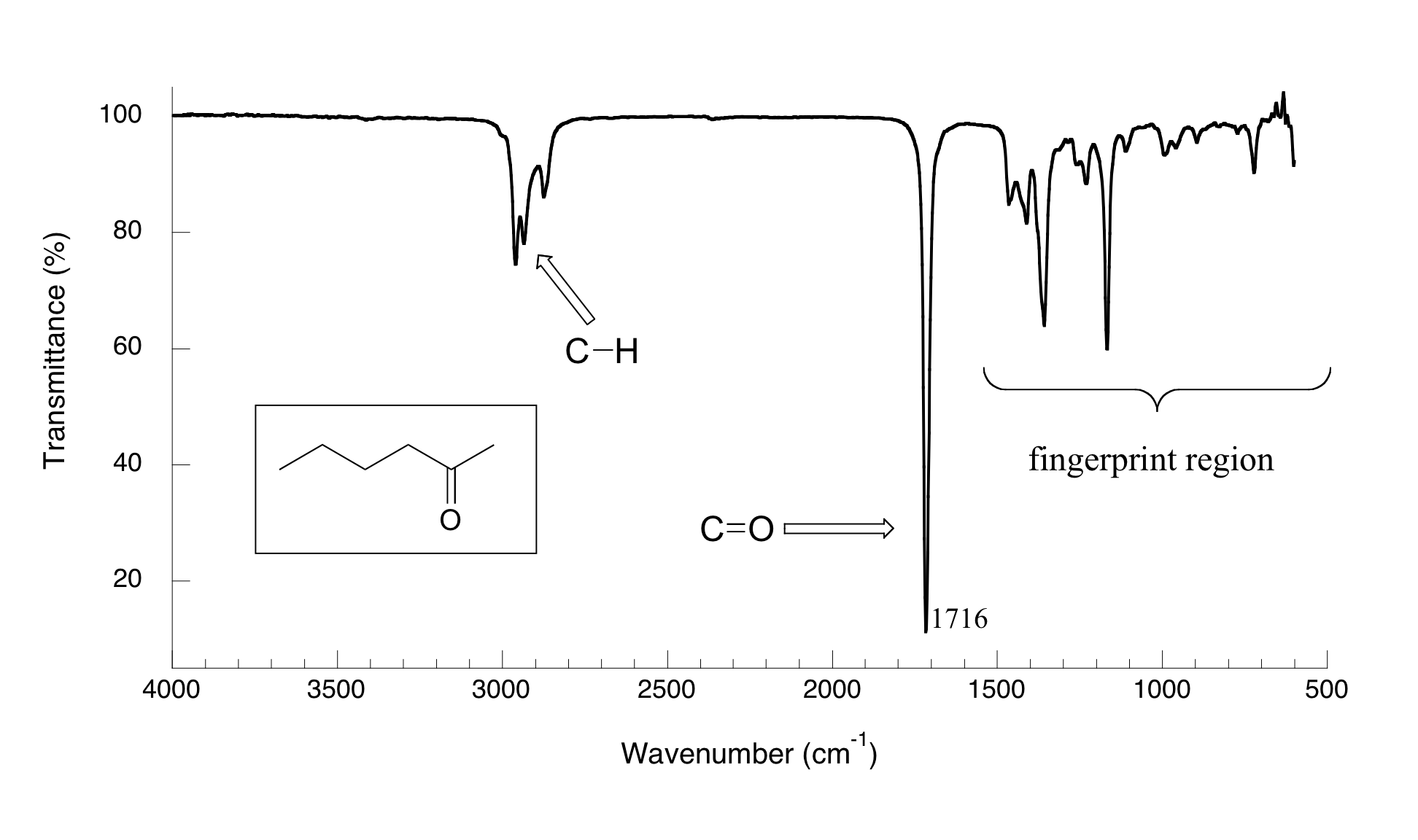
There are a number of things that need to be explained in order for you to understand what it is that we are looking at. On the horizontal axis we see IR wavelengths expressed in terms of a unit called wavenumber (cm-1), which tells us how many waves fit into one centimeter. On the vertical axis we see ‘% transmittance’, which tells us how strongly light was absorbed at each frequency (100% transmittance means no absorption occurred at that frequency). The solid line traces the values of % transmittance for every wavelength – the ‘peaks’ (which are actually pointing down) show regions of strong absorption. For some reason, it is typical in IR spectroscopy to report wavenumber values rather than wavelength (in meters) or frequency (in Hz). The ‘upside down’ vertical axis, with absorbance peaks pointing down rather than up, is also a curious convention in IR spectroscopy. We wouldn’t want to make things too easy for you!
After completing this section, you should be able to
- describe how the so-called “fingerprint region” of an infrared spectrum can assist in the identification of an unknown compound.
- identify the functional group or groups present in a compound, given a list of the most prominent absorptions in the infrared spectrum and a table of characteristic absorption frequencies.
- identify the broad regions of the infrared spectrum in which occur absorptions caused by
- \(\ce{\sf{N-H}}\), \(\ce{\sf{C-H}}\), and \(\ce{\sf{O-H}}\)
- \(\ce{\sf{C#C}}\) and \(\ce{\sf{C#N}}\)
- \(\ce{\sf{C=O}}\), \(\ce{\sf{C=N}}$, and $\ce{\sf{C=C}}\)
Make certain that you can define, and use in context, the key term below.
- fingerprint region
When answering assignment questions, you may use this IR table to find the characteristic infrared absorptions of the various functional groups. However, you should be able to indicate in broad terms where certain characteristic absorptions occur. You can achieve this objective by memorizing the following table.
| Region of Spectrum (cm−1) | Absorption |
|---|---|
| 2500-4000 | $\ce{\sf{N−H}}$, $\ce{\sf{O−H}}$, $\ce{\sf{C−H}}$ |
| 2000-2500 | $\ce{\sf{C#C}}$, $\ce{\sf{C#N}}$ |
| 1500-2000 | $\ce{\sf{C=O}}$, $\ce{\sf{C=N}}$, $\ce{\sf{C=C}}$ |
| below 1500 | Fingerprint region |
The Origin of Group Frequencies
An important observation made by early researchers is that many functional group absorb infrared radiation at about the same wavenumber, regardless of the structure of the rest of the molecule. For example, C-H stretching vibrations usually appear between 3200 and 2800cm-1 and carbonyl(C=O) stretching vibrations usually appear between 1800 and 1600cm-1. This makes these bands diagnostic markers for the presence of a functional group in a sample. These types of infrared bands are called group frequencies because they tell us about the presence or absence of specific functional groups in a sample.
.gif?revision=1&size=bestfit&width=720&height=335)
The region of the infrared spectrum from 1200 to 700 cm-1 is called the fingerprint region. This region is notable for the large number of infrared bands that are found there. Many different vibrations, including C-O, C-C and C-N single bond stretches, C-H bending vibrations, and some bands due to benzene rings are found in this region. The fingerprint region is often the most complex and confusing region to interpret, and is usually the last section of a spectrum to be interpreted. However, the utility of the fingerprint region is that the many bands there provide a fingerprint for a molecule.
The key absorption peak in this spectrum is that from the carbonyl double bond, at 1716 cm-1 (corresponding to a wavelength of 5.86 mm, a frequency of 5.15 x 1013 Hz, and a ΔE value of 4.91 kcal/mol). Notice how strong this peak is, relative to the others on the spectrum: a strong peak in the 1650-1750 cm-1 region is a dead giveaway for the presence of a carbonyl group. Within that range, carboxylic acids, esters, ketones, and aldehydes tend to absorb in the shorter wavelength end (1700-1750 cm-1), while conjugated unsaturated ketones and amides tend to absorb on the longer wavelength end (1650-1700 cm-1).
The jagged peak at approximately 2900-3000 cm-1 is characteristic of tetrahedral carbon-hydrogen bonds. This peak is not terribly useful, as just about every organic molecule that you will have occasion to analyze has these bonds. Nevertheless, it can serve as a familiar reference point to orient yourself in a spectrum.
You will notice that there are many additional peaks in this spectrum in the longer-wavelength 400 -1400 cm-1 region. This part of the spectrum is called the fingerprint region. While it is usually very difficult to pick out any specific functional group identifications from this region, it does, nevertheless, contain valuable information. The reason for this is suggested by the name: just like a human fingerprint, the pattern of absorbance peaks in the fingerprint region is unique to every molecule, meaning that the data from an unknown sample can be compared to the IR spectra of known standards in order to make a positive identification. In the mid-1990's, for example, several paintings were identified as forgeries because scientists were able to identify the IR footprint region of red and yellow pigment compounds that would not have been available to the artist who supposedly created the painting (for more details see Chemical and Engineering News, Sept 10, 2007, p. 28).
Now, let’s take a look at the IR spectrum for 1-hexanol.
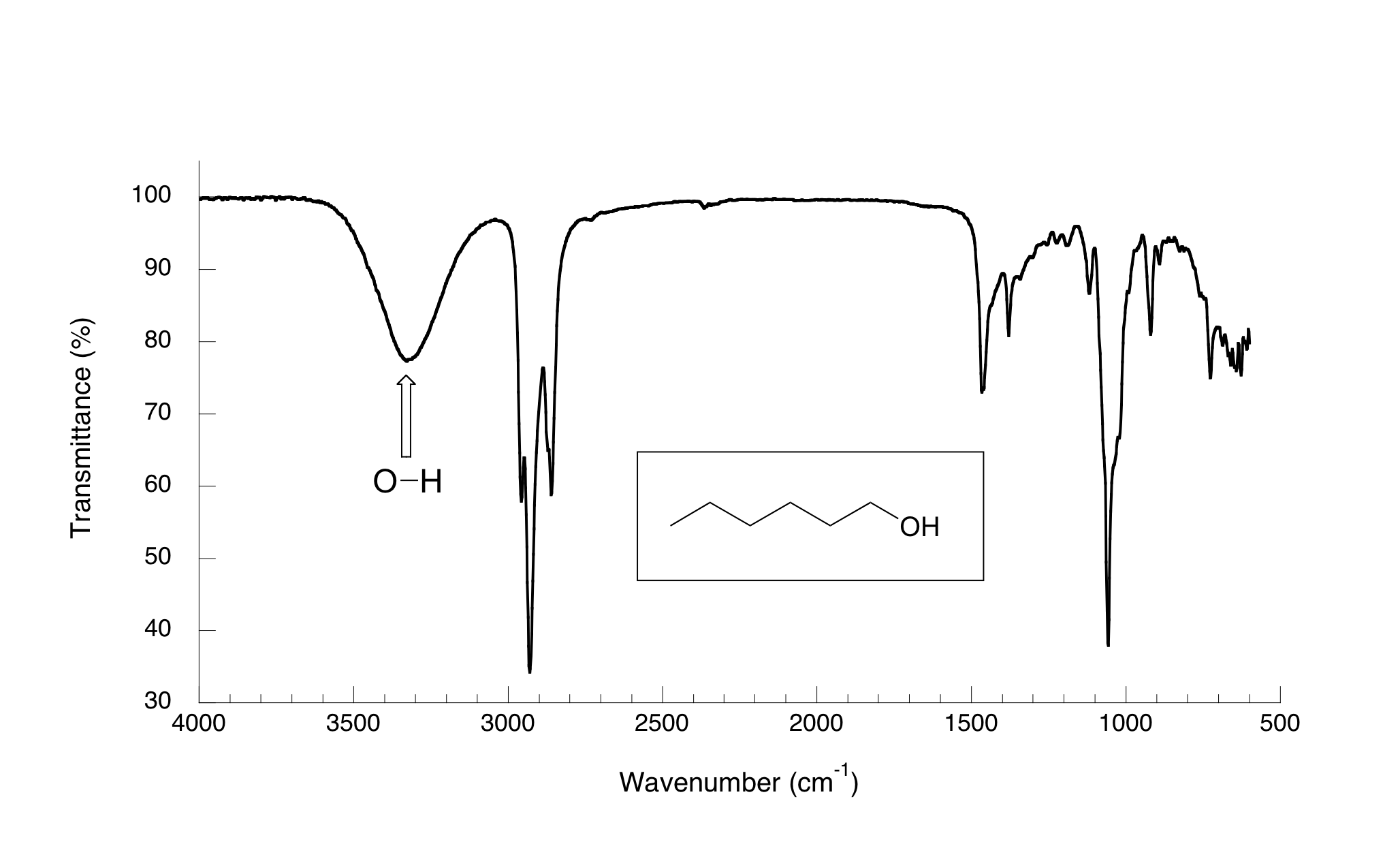
As you can see, the carbonyl peak is gone, and in its place is a very broad ‘mountain’ centered at about 3400 cm-1. This signal is characteristic of the O-H stretching mode of alcohols, and is a dead giveaway for the presence of an alcohol group. The breadth of this signal is a consequence of hydrogen bonding between molecules.
In the spectrum of octanoic acid we see, as expected, the characteristic carbonyl peak, this time at 1709 cm-1.

We also see a low, broad absorbance band that looks like an alcohol, except that it is displaced slightly to the right (long-wavelength) side of the spectrum, causing it to overlap to some degree with the C-H region. This is the characteristic carboxylic acid O-H single bond stretching absorbance.
The spectrum for 1-octene shows two peaks that are characteristic of alkenes: the one at 1642 cm-1 is due to stretching of the carbon-carbon double bond, and the one at 3079 cm-1 is due to stretching of the s bond between the alkene carbons and their attached hydrogens.
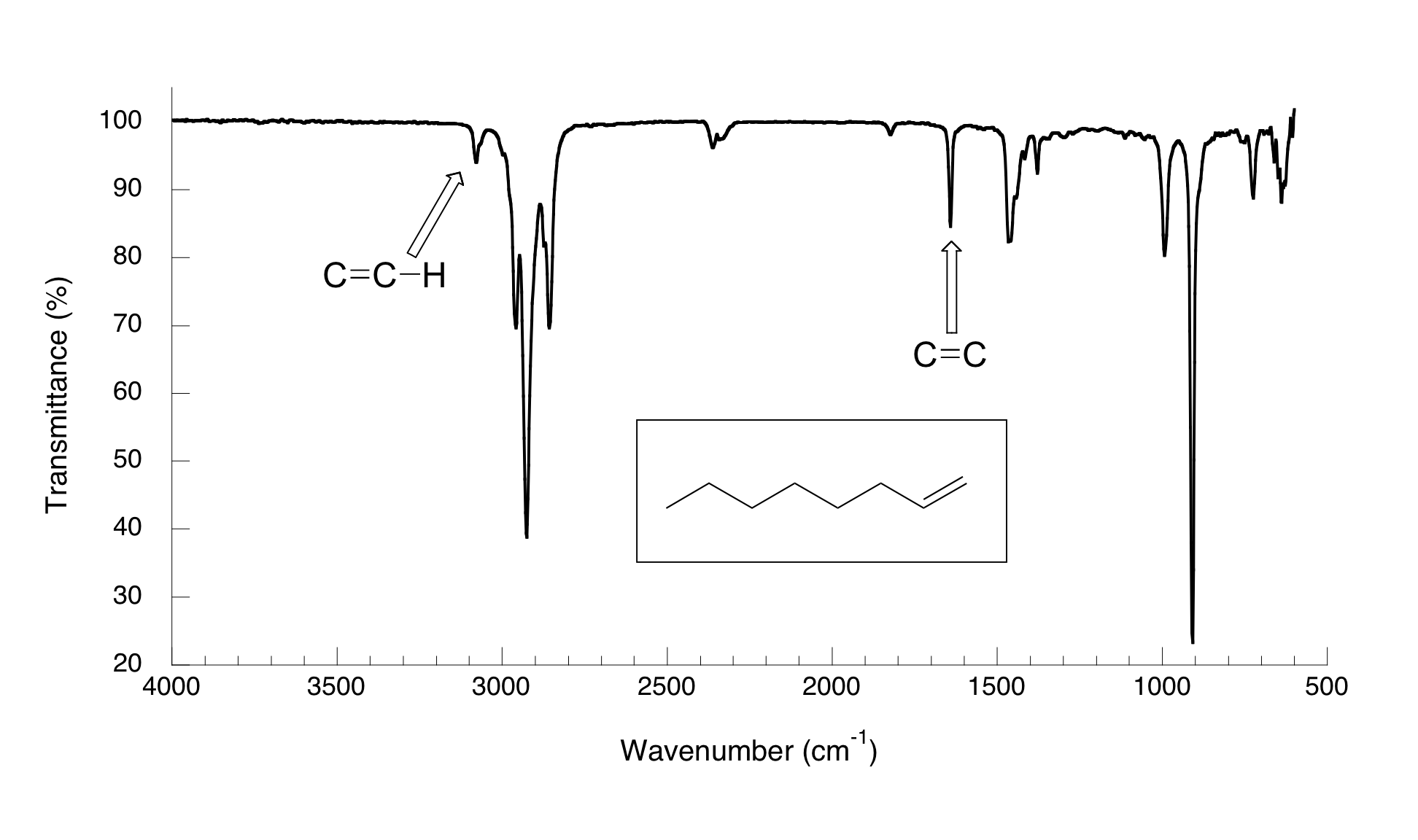
Alkynes have characteristic IR absorbance peaks in the range of 2100-2250 cm-1 due to stretching of the carbon-carbon triple bond, and terminal alkenes can be identified by their absorbance at about 3300 cm-1, due to stretching of the bond between the sp-hybridized carbon and the terminal hydrogen.
It is possible to identify other functional groups such as amines and ethers, but the characteristic peaks for these groups are considerably more subtle and/or variable, and often are overlapped with peaks from the fingerprint region. For this reason, we will limit our discussion here to the most easily recognized functional groups, which are summarized in this table.
As you can imagine, obtaining an IR spectrum for a compound will not allow us to figure out the complete structure of even a simple molecule, unless we happen to have a reference spectrum for comparison. In conjunction with other analytical methods, however, IR spectroscopy can prove to be a very valuable tool, given the information it provides about the presence or absence of key functional groups. IR can also be a quick and convenient way for a chemist to check to see if a reaction has proceeded as planned. If we were to run a reaction in which we wished to convert cyclohexanone to cyclohexanol, for example, a quick comparison of the IR spectra of starting compound and product would tell us if we had successfully converted the ketone group to an alcohol.
More examples of IR spectra
To illustrate the usefulness of infrared absorption spectra, examples for five C4H8O isomers are presented below their corresponding structural formulas. Try to associate each spectrum with one of the isomers in the row above it.
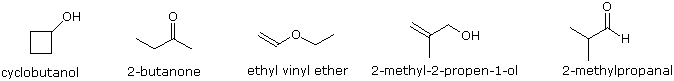
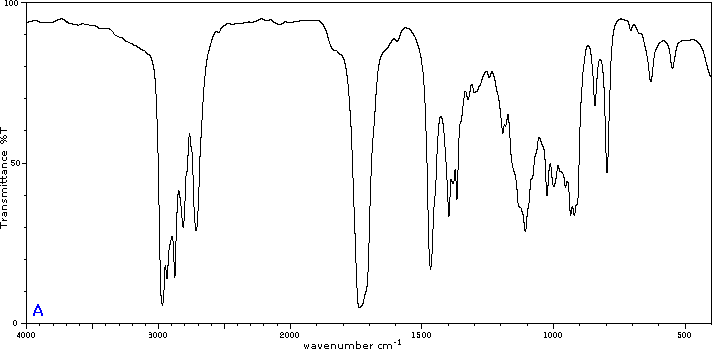
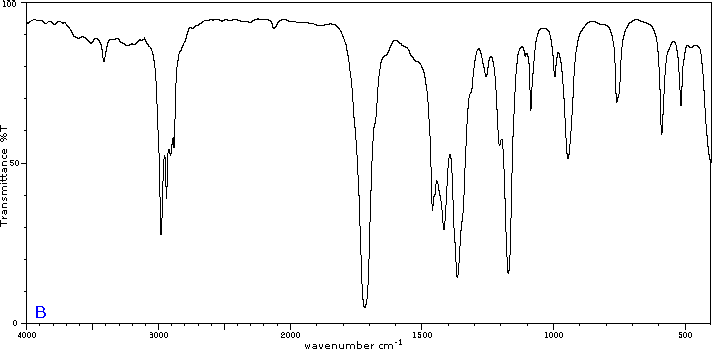
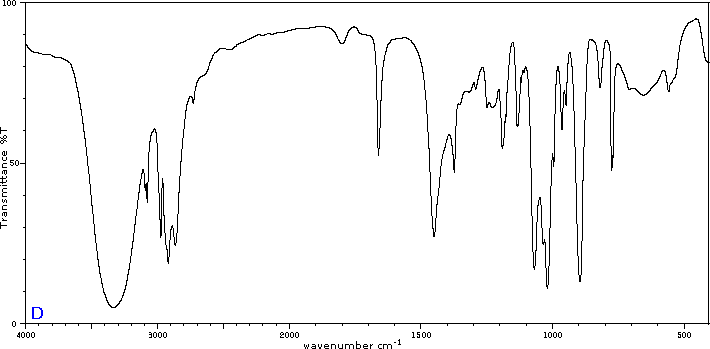
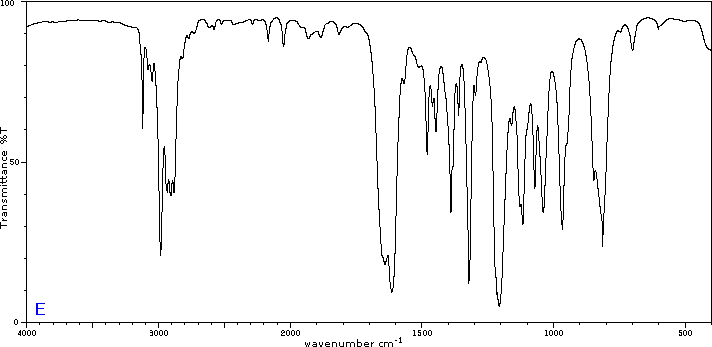
Exercises
After completing this section, you should be able to use an infrared spectrum to determine the presence of functional groups, such as alcohols, amines and carbonyl groups, in an unknown compound, given a list of infrared absorption frequencies.
In Chapter 12.7 you should have learned, in broad terms, where a few key absorptions occur. Otherwise, to find the characteristic infrared absorptions of the various functional groups, refer to this IR table.
Spectral Interpretation by Application of Group Frequencies
One of the most common application of infrared spectroscopy is to the identification of organic compounds. The major classes of organic molecules are shown in this category and also linked on the bottom page for the number of collections of spectral information regarding organic molecules.
Hydrocarbons
Hydrocarbons compounds contain only C-H and C-C bonds, but there is plenty of information to be obtained from the infrared spectra arising from C-H stretching and C-H bending.
In alkanes, which have very few bands, each band in the spectrum can be assigned:
- C–H stretch from 3000–2850 cm-1
- C–H bend or scissoring from 1470-1450 cm-1
- C–H rock, methyl from 1370-1350 cm-1
- C–H rock, methyl, seen only in long chain alkanes, from 725-720 cm-1
Figure 3. shows the IR spectrum of octane. Since most organic compounds have these features, these C-H vibrations are usually not noted when interpreting a routine IR spectrum. Note that the change in dipole moment with respect to distance for the C-H stretching is greater than that for others shown, which is why the C-H stretch band is the more intense.
.png?revision=1&size=bestfit&width=548&height=282)
In alkenes compounds, each band in the spectrum can be assigned:
- C=C stretch from 1680-1640 cm-1
- =C–H stretch from 3100-3000 cm-1
- =C–H bend from 1000-650 cm-1
Figure 4. shows the IR spectrum of 1-octene. As alkanes compounds, these bands are not specific and are generally not noted because they are present in almost all organic molecules.
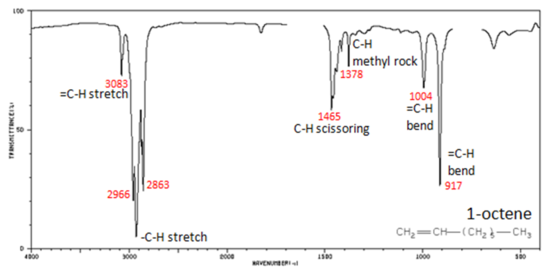
In alkynes, each band in the spectrum can be assigned:
- –C≡C– stretch from 2260-2100 cm-1
- –C≡C–H: C–H stretch from 3330-3270 cm-1
- –C≡C–H: C–H bend from 700-610 cm-1
The spectrum of 1-hexyne, a terminal alkyne, is shown below.
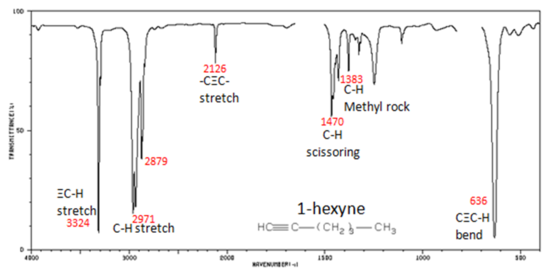
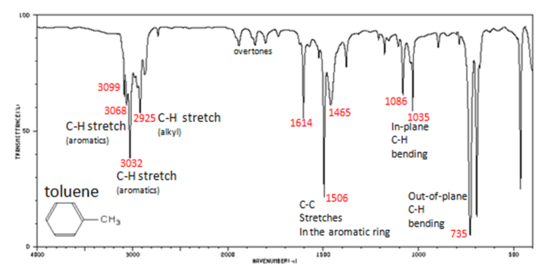
In aromatic compounds, each band in the spectrum can be assigned:
- C–H stretch from 3100-3000 cm-1
- overtones, weak, from 2000-1665 cm-1
- C–C stretch (in-ring) from 1600-1585 cm-1
- C–C stretch (in-ring) from 1500-1400 cm-1
- C–H "oop" from 900-675 cm-1
Note that this is at slightly higher frequency than is the –C–H stretch in alkanes. This is a very useful tool for interpreting IR spectra. Only alkenes and aromatics show a C–H stretch slightly higher than 3000 cm-1.
Figure 6. shows the spectrum of toluene.
Functional Groups Containing the C-O Bond
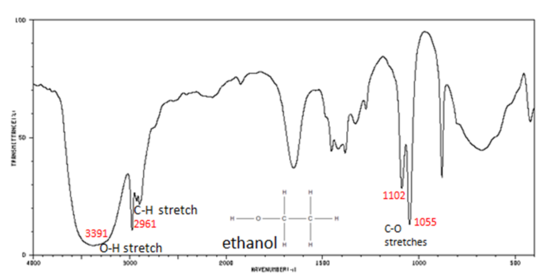
Alcohols have IR absorptions associated with both the O-H and the C-O stretching vibrations.
- O–H stretch, hydrogen bonded 3500-3200 cm-1
- C–O stretch 1260-1050 cm-1 (s)
Figure 7. shows the spectrum of ethanol. Note the very broad, strong band of the O–H stretch.
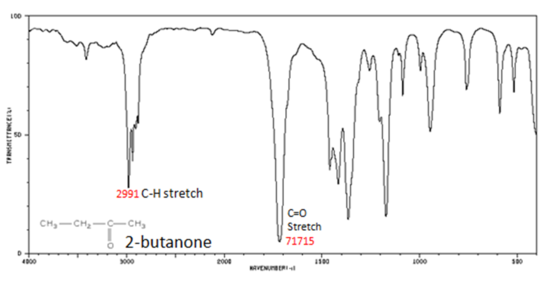
The carbonyl stretching vibration band C=O of saturated aliphatic ketones appears:
- C=O stretch - aliphatic ketones 1715 cm-1
- α, β -unsaturated ketones 1685-1666 cm-1
Figure 8. shows the spectrum of 2-butanone. This is a saturated ketone, and the C=O band appears at 1715.
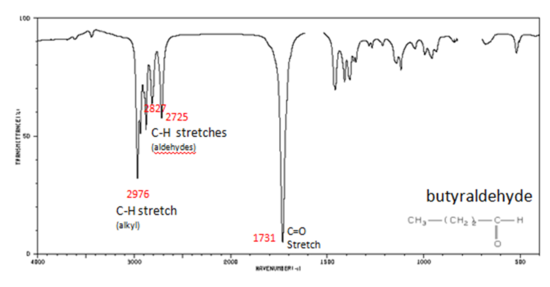
If a compound is suspected to be an aldehyde, a peak always appears around 2720 cm-1 which often appears as a shoulder-type peak just to the right of the alkyl C–H stretches.
- H–C=O stretch 2830-2695 cm-1
- C=O stretch:
- aliphatic aldehydes 1740-1720 cm-1
- α, β -unsaturated aldehydes 1710-1685 cm-1
Figure 9. shows the spectrum of butyraldehyde.
The carbonyl stretch C=O of esters appears:
- C=O stretch
- aliphatic from 1750-1735 cm-1
- α, β -unsaturated from 1730-1715 cm-1
- C–O stretch from 1300-1000 cm-1
Figure 10. shows the spectrum of ethyl benzoate.
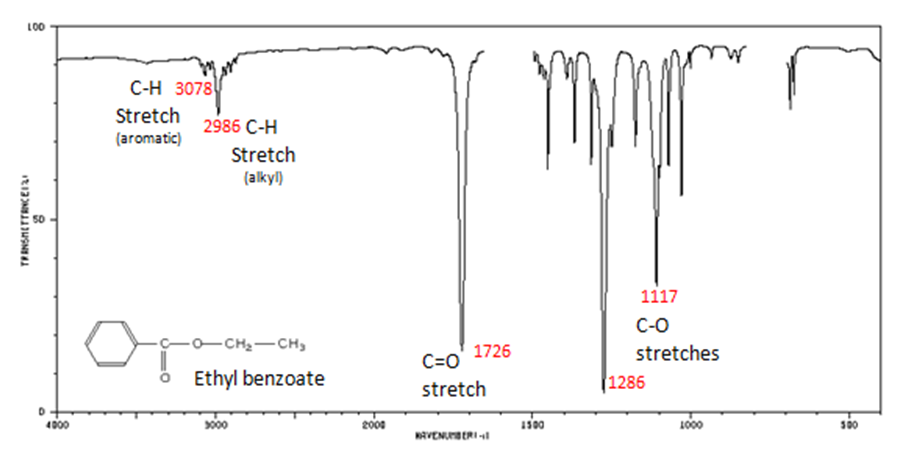
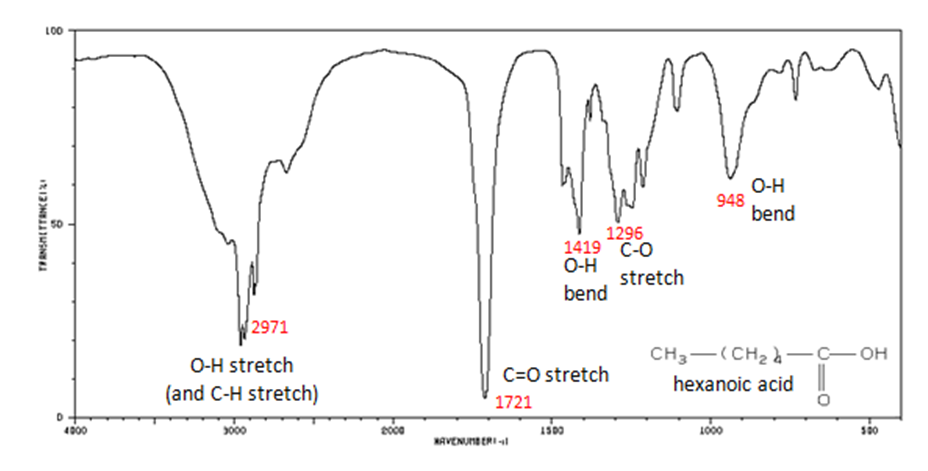
The carbonyl stretch C=O of a carboxylic acid appears as an intense band from 1760-1690 cm-1. The exact position of this broad band depends on whether the carboxylic acid is saturated or unsaturated, dimerized, or has internal hydrogen bonding.
- O–H stretch from 3300-2500 cm-1
- C=O stretch from 1760-1690 cm-1
- C–O stretch from 1320-1210 cm-1
- O–H bend from 1440-1395 and 950-910 cm-1
Figure 11. shows the spectrum of hexanoic acid.
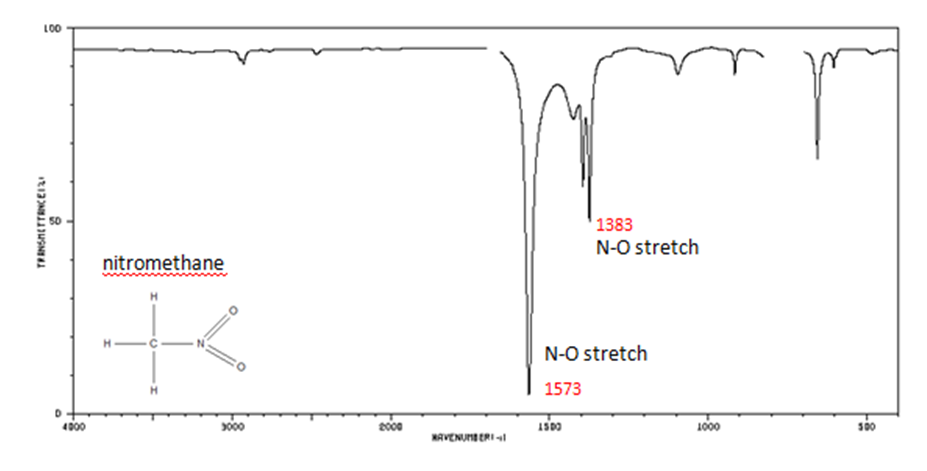
Organic Nitrogen Compounds
- N–O asymmetric stretch from 1550-1475 cm-1
- N–O symmetric stretch from 1360-1290 cm-1
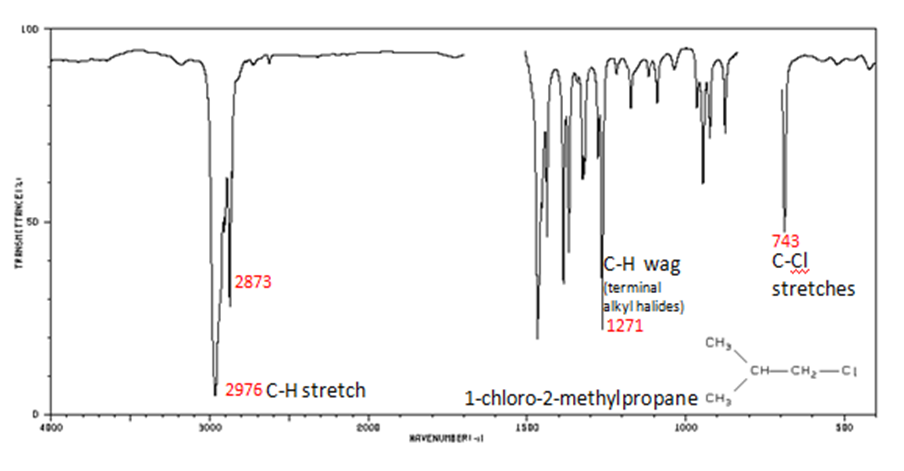
Organic Compounds Containing Halogens
Alkyl halides are compounds that have a C–X bond, where X is a halogen: bromine, chlorine, fluorene, or iodine.
- C–H wag (-CH2X) from 1300-1150 cm-1
- C–X stretches (general) from 850-515 cm-1
- C–Cl stretch 850-550 cm-1
- C–Br stretch 690-515 cm-1
The spectrum of 1-chloro-2-methylpropane are shown below.
For more Infrared spectra Spectral database of organic molecules is introduced to use free database. Also, the infrared spectroscopy correlation table is linked on bottom of page to find other assigned IR peaks.
Exercises
Caffeine has a mass of 194.19 amu, determined by mass spectrometry, and contains C, N, H, O. What is a molecular formula for this molecule?
- Answer
-
C8H10N4O2
C = 12 × 8 = 96
N = 14 × 4 = 56
H = 1 × 10 = 10
O = 2 × 16 = 32
96+56+10+32 = 194 g/mol
The following are the spectra for 2-methyl-2-hexene and 2-heptene, which spectra belongs to the correct molecule. Explain.
A:
B:
Source: SDBSWeb : http://sdbs.db.aist.go.jp (National Institute of Advanced Industrial Science and Technology, 2 December 2016)
- Answer
-
The (A) spectrum is 2-methyl-2-hexene and the (B) spectrum is 2-heptene. Looking at (A) the peak at 68 m/z is the fractioned molecule with just the tri-substituted alkene present. While (B) has a strong peak around the 56 m/z, which in this case is the di-substituted alkene left behind from the linear heptene.
What are the masses of all the components in the following fragmentations?
- Answer
-
Which of the following frequencies/wavelengths are higher energy
A. λ = 2.0x10-6 m or λ = 3.0x10-9 m
B. υ = 3.0x109 Hz or υ = 3.0x10-6 Hz
- Answer
-
A. λ = 3.0x10-9 m
B. υ = 3.0x109 Hz
Calculate the energies for the following;
A. Gamma Ray λ = 4.0x10-11 m
B. X-Ray λ = 4.0x10-9 m
C. UV light υ = 5.0x1015 Hz
D. Infrared Radiation λ = 3.0x10-5 m
E. Microwave Radiation υ = 3.0x1011 Hz
- Answer
-
A. 4.965x10-15 J
B. 4.965x10-17 J
C. 3.31x10-18 J
D. 6.62x10-21 J
E. 1.99x10-22 J
What functional groups give the following signals in an IR spectrum?
A) 1700 cm-1
B) 1550 cm-1
C) 1700 cm-1 and 2510-3000 cm-1
- Answer
-
How can you distinguish the following pairs of compounds through IR analysis?
A) CH3OH (Methanol) and CH3CH2OCH2CH3 (Diethylether)
B) Cyclopentane and 1-pentene.
C)
- Answer
-
A) A OH peak will be present around 3300 cm-1 for methanol and will be absent in the ether.
B) 1-pentene will have a alkene peak around 1650 cm-1 for the C=C and there will be another peak around 3100 cm-1 for the sp2 C-H group on the alkene
C) Cannot distinguish these two isomers. They both have the same functional groups and therefore would have the same peaks on an IR spectra.
The following spectra is for the accompanying compound. What are the peaks that you can I identify in the spectrum?
Source: SDBSWeb : http://sdbs.db.aist.go.jp (National Institute of Advanced Industrial Science and Technology, 2 December 2016)
- Answer
-
Frequency (cm-1) Functional Group
3200 C≡C-H
2900-3000 C-C-H, C=C-H
2100 C≡C
1610 C=C
(There is also an aromatic undertone region between 2000-1600 which describes the substitution on the phenyl ring.)
What absorptions would the following compounds have in an IR spectra?
- Answer
-
A)
Frequency (cm-1) Functional Group
2900-3000 C-C-H, C=C-H
1710 C=O
1610 C=C
1100 C-O
B)
Frequency (cm-1) Functional Group
3200 C≡C-H
2900-3000 C-C-H, C=C-H
2100 C≡C
1710 C=O
C)
Frequency (cm-1) Functional Group
3300 (broad) O-H
2900-3000 C-C-H, C=C-H
2000-1800 Aromatic Overtones
1710 C=O
1610 C=C