
4.4: Searching PubChem Using a Non-Textual Query
- Page ID
- 170162
This section describes various searches that can be performed in PubChem.1,2,3 Currently PubChem has three different search interfaces:
- PubChem homepage (http://pubchem.ncbi.nlm.nih.gov)
- PubChem Chemical Structure Search (https://pubchem.ncbi.nlm.nih.gov/search/search.cgi)
- PubChem Search (https://pubchem.ncbi.nlm.nih.gov/search/).
As explained in Section 4.1, the PubChem homepage provides a search interface for all three primary databases (e.g., Substance, Compound, and BioAssay). However, the search box on the PubChem homepage can accepts textual keywords only, and it is difficult to input non-textual queries (such as chemical structures). The PubChem Chemical Structure Search allows users to perform various searches using both textual and non-textual queries. This search interface is integrated with PubChem Sketcher,4 which enables users to provide the 2-D structure of a molecule as a query for chemical structure search. While the PubChem Chemical Structure Search is limited to search for chemical structures, the PubChem Search allows users to search for bioassays, bioactivities, patents, and targets as well as chemical structures, but it is still in beta testing. In this module, we use the Chemical Structure Search for chemical structure search.
Molecular formula search
Molecular formula search allows one to find molecules that contain a certain number and type of elements. Typically, molecular formula search returns by default molecules that exactly match the queried stoichiometry. For example, a query of “C6H6” will return all structures containing six carbon atoms, six hydrogen atoms and nothing else. However, molecular formula search implemented in some databases, including PubChem Chemical Structure Search, has an option to allow other elements in returned hits (e.g., C6H6O or C6H6N2O for the “C6H6” query).
Identity search
Identity search is to locate a particular chemical structure that is “identical” to the query chemical structure. Although identity search seems conceptually straightforward, one should keep in mind that the word “identical” can have different notions. For example, if a molecule exists as multiple tautomeric forms in equilibrium, do you want to consider all these tautomers identical and search the database for all of them? If your query molecule has a chiral stereo center, should you consider both R- and S-forms in your search? In your identity search, do you want to include isotopically substituted species of the provided query molecule as well as the query itself? Depending on how to deal with these nuances of chemical structures, identical search will return different results. The identity search in the PubChem Chemical Structure Search allows users to choose a desired degree of “sameness” from several predefined options. To see these options, one need to expand the options section by clicking the “plus” button next to the “option” section heading.
Substructure and superstructure search
When a chemical structure occurs as a part of a bigger chemical structure, the former is called a substructure and the latter is referred to as a superstructure. For example, ethanol is a substructure of acetic acid, and acetic acid is a superstructure of ethanol.
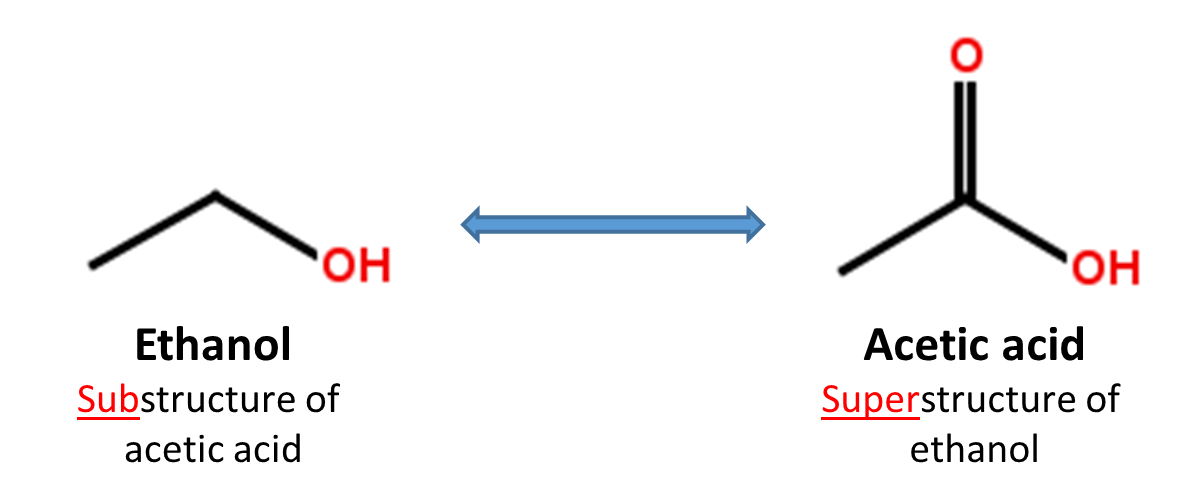
In substructure search, one provides an input substructure as a query to find molecules that contain the query substructure (that is, superstructures that contain the query substructure). On the contrary, superstructure search returns molecules that comprise or make up the provided chemical structure query (that is, substructures that is contained in the query superstructure). It should be noted that substructure search does not give you substructures of the query and that superstructure search does not return superstructures of the query.
It is possible to include explicit hydrogen atoms as part of the pattern being searched. For example, if you choose to do so, the SMILES queries [CH2][CH2][OH] and [CH3][CH][OH] will return molecules whose formula are R-CH2-CH2-OH and CH3-CH(R)-OH, respectively. Substructure/superstructure searches implemented in some databases remove by default explicit hydrogens from the query molecule prior to search, the two SMILES queries [CH2][CH2][OH] and [CH3][CH][OH] may give you the same result as what the SMILES query CCO does, unless you specify that explicit hydrogens should be included in pattern matching.
In addition to explicit hydrogen atoms, there are additional factors that may affect results of substructure/superstructure searches, for example, whether to ignore stereochemistry, isotopism, tautomerism, formal charge, and so on.
Similarity search
Molecular similarity (also called chemical similarity or chemical structure similarity) is a fundamental concept in cheminformatics, playing an important role in computational methods for predicting properties of chemical compounds as well as designing chemicals with desired properties. The underlying assumption in these computational methods is that structurally similar molecules are likely to have similar biological and physicochemical properties (commonly called the similarity principle).5 Molecular similarity is a straightforward and easy-to-understand concept, but there is no absolute, mathematical definition of molecular similarity that everyone agrees on. As a result, there are a virtually infinite number of molecular similarity methods, which quantify molecular similarity. Similarity search uses a molecular similarity method to find molecules similar to the query structure.
Two-dimensional (2-D) similarity methods
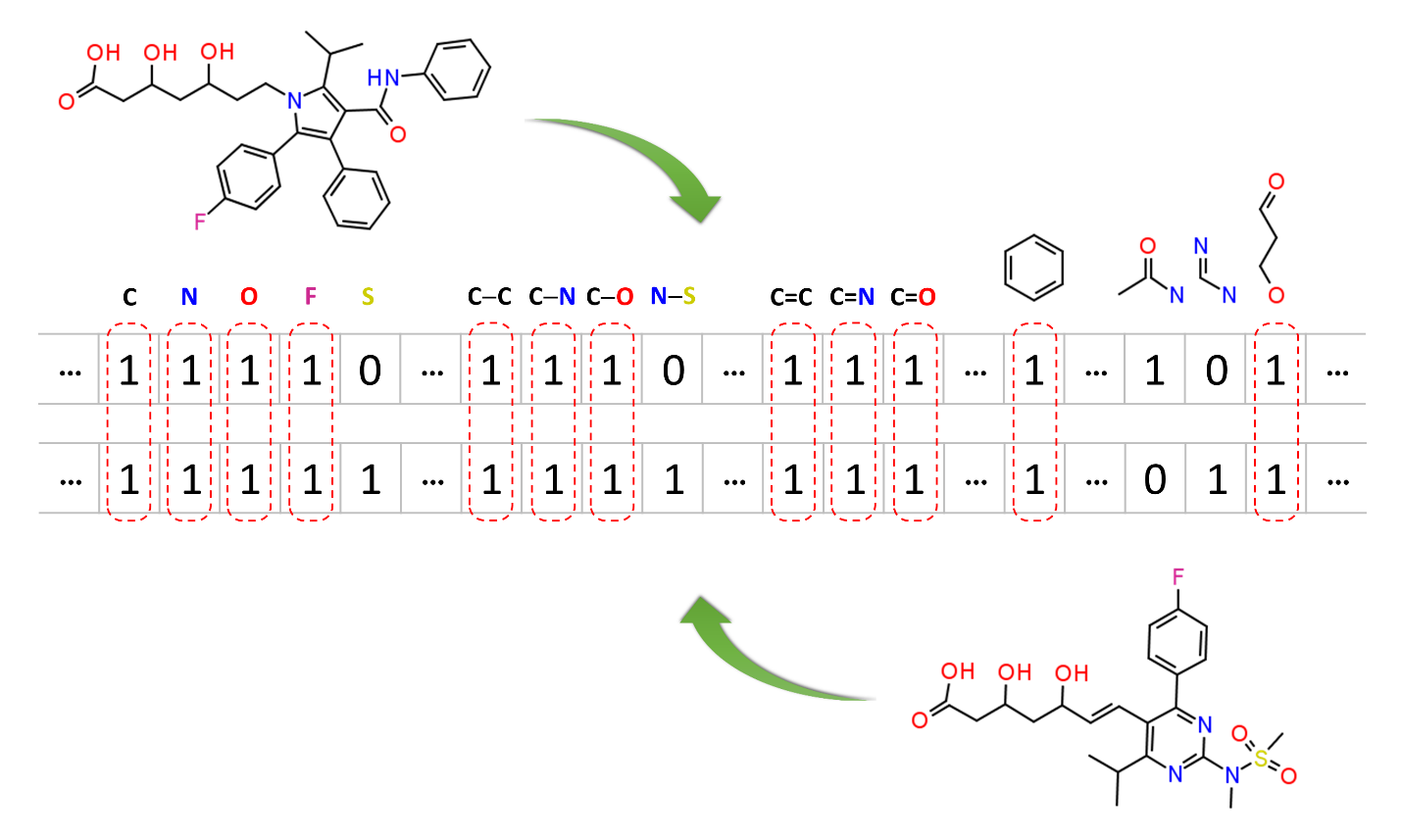
Molecular similarity methods can be broadly classified into two-dimensional (2-D) and three-dimensional (3-D) similarity methods. Typically, 2-D similarity methods use so-called molecular fingerprints. The most common types of molecular fingerprints are structural keys, which encode structural information of a molecule into a binary string (that is, a string of 0’s and 1’s). The position of each number in this string corresponds to a particular fragment. If the molecule has a particular fragment, the corresponding bit position is set to 1, and otherwise to 0. Note that there are many different ways to design molecular fingerprints, depending on what fragments are included in the fingerprint definition. PubChem uses its own fingerprint called PubChem subgraph fingerprints.
In 2-D similarity methods, structural similarity between two molecules is estimated by comparing their molecular fingerprints. Their similarity is quantified as a so-called similarity score or similarity coefficient. While several different methods can be used for computation of a similarity score, the underlying ideas are the same as each other: if the two fingerprints have 1’s at the same position, it means that both compounds have the same fragment, and if the molecules share more common fragments, they are considered to be more similar. In conjunction with the PubChem subgraph fingerprints, PubChem 2-D similarity method use the Tanimoto coefficient6-8
\[Tanimoto=\frac{N_{AB}}{N_A+N_B-N_{AB}}\label{Einstein}\]
where NA and NB are the number of bits set in the fingerprints for molecules A and B, respectively, and NAB is the number of bits set in both fingerprints. The Tanimoto score ranges from 0 (for no similarity) to 1 (for identical molecules). 2-D Similarity search returns molecules whose similarity scores with the query molecule are greater than or equal to a given Tanimoto cut-off value.
PubChem 3-D similarity method
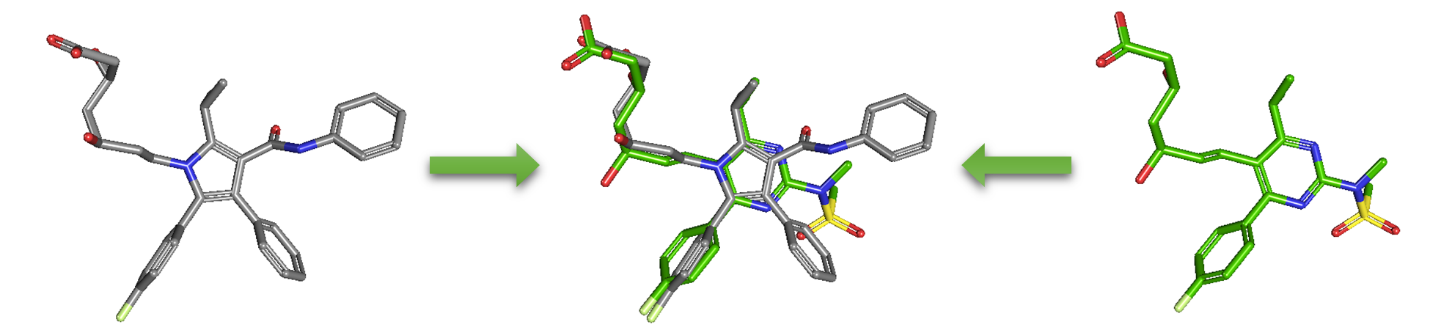
As an alternative to 2-D similarity search, 3-D similarity search can also be performed using the “3D conformer” tab in PubChem Chemical Structure Search. 3-D similarity methods use the 3-D structures (that is, conformations) of molecules. PubChem’s 3-D similarity method is based on the atom-centered Gaussian-shape comparison method by Grant and coworkers,9,10,11,12 implemented in the Rapid Overlay of Chemical Structures (ROCS).13,14 While the underlying mathematics of this approach is beyond the scope of this module, what this method essentially does is to find the “best” alignment of the 3-D structures of two molecules, which gives the maximized overlap between them. The 3-D similarity method quantifies the 3-D molecular similarity using three metrics.
- Shape-Tanimoto (ST): quantifies steric shape similarity between two conformers.
- Color-Tanimoto (CT): quantifies the overlap of functional groups between two conformers, such as hydrogen bond donors and acceptors, cations, anions, rings, and hydrophobes.
- Combo-Tanimoto (ComboT): the sum of ST and CT scores between two conformers. It takes into account the shape similarity (ST) and functional group similarity (CT) simultaneously.
Because both the ST and CT scores range from 0 (for no similarity) to 1 (for identical molecules), the ComboT score may have a value from 0 to 2 (without normalization to unity). Note that the ST, CT and ComboT scores between two molecules can be evaluated in two different molecular superpositions: (1) in the ST- or shape-optimized superpositions, and (2) in the CT- or feature-optimization superpositions. In the ST-optimization approach, the shape overlap between the molecules (that is, the ST score) are maximized and the single-point CT score is evaluated at that superposition. On the contrary, the CT-optimization considers both ST and CT scores to find the best superposition between molecules, and the single-point ST score is computed at that superposition.
The 3-D similarity method used in PubChem requires the 3-D structures of molecules. PubChem generates a conformer ensemble containing up to 500 conformers for each compound that satisfy the following conditions15,16,17:
- Not too big or too flexible (with £ 50 non-hydrogen atoms and ≤ 15 rotatable bonds).
- Have only a single covalent unit (i.e., not a salt or a mixture).
- Consist of only supported elements (H, C, N, O, F, Si, P, S, Cl, Br, and I).
- Contain only atom types recognized by the MMFF94s force field.
- Fewer than six undefined atom or bond stereo centers.
About 90% of compounds in PubChem have computationally generated conformer models. Although each compound has up to 500 conformers (depending on the molecular size and flexibility), many PubChem tools and services support up to 10 conformers per compound. It should be emphasized that these conformers are not energy-minimized but sampled from the conformational space of a given molecule in such a way that the sampled conformers represent the overall diversity of shape and feature of the molecule.15,16,17 These conformer models aim to generate bioactive conformers, which would be found in protein-ligand complexes. For this reason, these conformers are often very different from their experimental structures determined in the gas phase.
References
(3) Kim, S. Expert Opinion on Drug Discovery 2016, 11, 843.
(4) Ihlenfeldt, W. D.; Bolton, E. E.; Bryant, S. H. J. Cheminform. 2009, 1, 20.
(6) Chen, X.; Reynolds, C. H. J. Chem. Inf. Comput. Sci. 2002, 42, 1407.
(8) Holliday, J. D.; Salim, N.; Whittle, M.; Willett, P. J. Chem. Inf. Comput. Sci. 2003, 43, 819.
(9) Grant, J. A.; Pickup, B. T. Journal of Physical Chemistry 1995, 99, 3503.
(10) Grant, J. A.; Gallardo, M. A.; Pickup, B. T. Journal of Computational Chemistry 1996, 17, 1653.
(11) Grant, J. A.; Pickup, B. T. Journal of Physical Chemistry 1996, 100, 2456.
(14) 3.1.0 ed.; OpenEye Scientific Software, Inc.: Santa Fe, NM, 2010.
(16) Bolton, E. E.; Kim, S.; Bryant, S. H. J. Cheminform. 2011, 3, 4.
(17) Kim, S.; Bolton, E. E.; Bryant, S. H. J. Cheminform. 2013, 5, 1.