
2.5: Structural Data Files
- Page ID
- 154857
- Gain understanding of chemical structural data files
- Survey data formats
- Survey molecular visualization and manipulation software and web services
Introduction
Structural data files are the files software agents typically use when processing chemical structural information, but can also contain additional information like molecular spectra. In principle you could say that there are two major components any structural data file, the simplified connection table and additional information. In effect the InChI line notation sort of models them, in that the main layer is the simplified connection table and the other layers are the additional information, except that in a structural data files hydrogen can be implicit or explicit (in the InChI they are explicit). So when you look at the different types of structural data files you will see they all have an atom table and a bond table. Information about individual atoms like isotopic definitions are associated with the atom table. That atom table may also indicate the 3d coordinates associated with a specific environment, and if that information is missing software agents will use an energy minimization calculation to determine 3D structure of an isolated atom.
In this section we will give a brief of the different types of structural data files and a survey of software programs and web services that can be used to display and manipulate structural data files, with a focus on open source options. There will be some overlap with these software programs and next section on chemical resolvers, which allow you to convert between file types.
Common Types of Structural Data Files
There are a variety of file formats and the most common are based on the MDL Molfile, of which V2000 is the most common, although V3000 is also commonly used. The SDF (Structure Data File) is based on the Molfile and figure represent an SDF file for acetone obtained through the NCI/CADD chemical identifier resolver.
Molfile
The following is a molfile for acetone obtained from the NCI chemical resolver. All molfiles have a header and a connection table (CTAB) that has two blocks, the Atom Block and the Bond Block.
The Header block has two lines, the first gives the name/formula of the molecule (if known) and is of variable format, the second gives the program that made file, the date and time it was made, and if 2D or 3D coordintates are given (figure 2.5.1 was created June 5, 2019 at 22:46 and has 3D coordinates).
The Count line block tells us acetone has 10 atoms and 9 bonds, it also provides the version number of the molfile. N
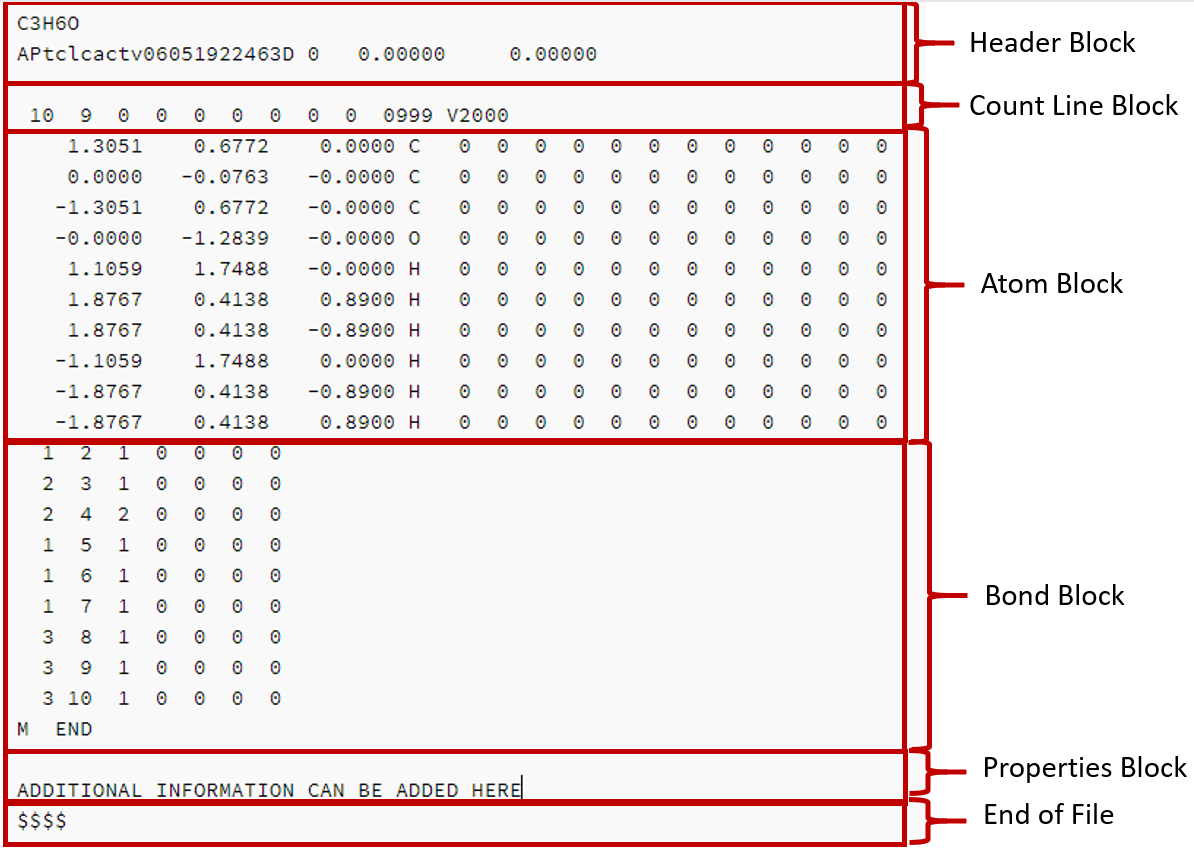
Figure \(\PageIndex{1}\): Molefile for acetone
Go to the NCI Chemical Identifier Resolver (https://cactus.nci.nih.gov/chemical/structure ); in Structure Identifier type "acetone", choose convert to SD File and submit.
Compare your file to figure 2.5.1, and hopefully the only difference you will see is the date the file was generated. We will discuss chemical resolvers in the next section.
Professor Bob Hanson at Saint Olaf College created a program for an earlier offering of this class called Hack-a-Mol that we will use to explore data files.
https://chemapps.stolaf.edu/jmol/jsmol/hackamol.htm
Open Hack-a-mol in a new window and search NCI for "acetone".
Now compare the molfile to the molfile from activity 2.5.1. What is the difference, and can you explain what is going on?
Open a new browser window, load Hack-a-Mol, search for Acetone, but use Pubchem instead of NCI.
Note the atom numbers in the atom block are implicit starting with 1 and going down to 10. We can also see that the oxygen is atom 4. From the bond block we see that atom 4 is attached to atom 2 (carbon) and it is a double bond. We also see that atom two is involved with two additional bonds, one to atom 1 and the other to atom 3, and both of those atoms are carbon. The connection table defines the molecules connectivity, and when coupled with 3D coordinates, gives its geometric shape. In this particular table we have included the hydrogens explicitly, but they could have been ommited. Also note the file ends with the four dollar signs, $$$$.
Figure 2.5.2 is the same data
C3H6O
APtclcactv06051922463D 0 0.00000 0.00000
10 9 0 0 0 0 0 0 0 0999 V2000
1.3051 0.6772 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 -0.0763 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.3051 0.6772 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 -1.2839 -0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1.1059 1.7488 -0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.8767 0.4138 0.8900 H 0 0 0 0 0 0 0 0 0 0 0 0
1.8767 0.4138 -0.8900 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.1059 1.7488 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.8767 0.4138 -0.8900 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.8767 0.4138 0.8900 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0 0
2 3 1 0 0 0 0
2 4 2 0 0 0 0
1 5 1 0 0 0 0
1 6 1 0 0 0 0
1 7 1 0 0 0 0
3 8 1 0 0 0 0
3 9 1 0 0 0 0
3 10 1 0 0 0 0
M END
ADDITIONAL INFORMATION CAN BE ADDED HERE
$$$$
180
-OEChem-06051922532D
10 9 0 0 0 0 0 0 0999 V2000
3.7320 0.7500 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8660 0.2500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0000 0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.8660 -0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3100 1.2869 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.4631 1.0600 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.6900 0.2131 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
2.2460 -0.7500 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
2.8660 -1.3700 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
3.4860 -0.7500 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0 0 0 0
2 3 1 0 0 0 0
2 4 1 0 0 0 0
3 5 1 0 0 0 0
3 6 1 0 0 0 0
3 7 1 0 0 0 0
4 8 1 0 0 0 0
4 9 1 0 0 0 0
4 10 1 0 0 0 0
M END
> <PUBCHEM_COMPOUND_CID>
180
> <PUBCHEM_COMPOUND_CANONICALIZED>
1
> <PUBCHEM_CACTVS_COMPLEXITY>
26.3
> <PUBCHEM_CACTVS_HBOND_ACCEPTOR>
1
> <PUBCHEM_CACTVS_HBOND_DONOR>
0
> <PUBCHEM_CACTVS_ROTATABLE_BOND>
0
> <PUBCHEM_CACTVS_SUBSKEYS>
AAADcYBAIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGgAAAAAACASAgAACAAAAAAAIAIAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA==
> <PUBCHEM_IUPAC_OPENEYE_NAME>
acetone
> <PUBCHEM_IUPAC_CAS_NAME>
2-propanone
> <PUBCHEM_IUPAC_NAME_MARKUP>
propan-2-one
> <PUBCHEM_IUPAC_NAME>
propan-2-one
> <PUBCHEM_IUPAC_SYSTEMATIC_NAME>
propan-2-one
> <PUBCHEM_IUPAC_TRADITIONAL_NAME>
acetone
> <PUBCHEM_IUPAC_INCHI>
InChI=1S/C3H6O/c1-3(2)4/h1-2H3
> <PUBCHEM_IUPAC_INCHIKEY>
CSCPPACGZOOCGX-UHFFFAOYSA-N
> <PUBCHEM_XLOGP3_AA>
-0.1
> <PUBCHEM_EXACT_MASS>
58.042
> <PUBCHEM_MOLECULAR_FORMULA>
C3H6O
> <PUBCHEM_MOLECULAR_WEIGHT>
58.08
> <PUBCHEM_OPENEYE_CAN_SMILES>
CC(=O)C
> <PUBCHEM_OPENEYE_ISO_SMILES>
CC(=O)C
> <PUBCHEM_CACTVS_TPSA>
17.1
> <PUBCHEM_MONOISOTOPIC_WEIGHT>
58.042
> <PUBCHEM_TOTAL_CHARGE>
Hack-a-Mol
Here’s a website at St. Olaf College where you can play with the relationship between 2D structures, 3D renderings, identifiers, and connection tables, courtesy of the cheminformatician Bob Hanson. There’s a link on the page to a document explaining “How it Works” (also linked here). As this course proceeds you will learn how we communicate with the NCI resolver and PubChem, and many of the fundamental features behind this application.
We have also embedded Hack-a-Mol below, and when doing your assignments you may want to open in a new window.
Hack-a-MolThis page is designed especially for students of cheminformatics who are just starting to learn about how chemical structures are represented digitally.With this page you can draw a structure in 2D, compare that with its 3D structure, and also see its structural data in a variety of formats. You can also enter a chemical identifier -- a chemical name, a SMILES string, or a Chemical Abstracts Registry Number, for instance -- in the box under the JSmol window. If you hack the structural data (carefully!) and then press ENTER, the 2D and 3D structures will update. You can also drag-drop a structure file into the JSmol window or copy/paste it into the textarea. How It Works Author: Bob Hanson |
|
labels console info clear no info |
|
| InChI: InChIKey: SMILES: |
|||
 Modify the data and press ENTER to see changes above. UNDO Modify the data and press ENTER to see changes above. UNDO |
|||
Let’s take another look at benzoic acid. Clear the 2D sketch window using the white box button at the top, second from the left, and then draw benzoic acid. Click the right arrow button. That should render a 3D structure in the window to the right and generate a MOL file in the text window below. (For details on how where this data comes from, see “2D to 3D” and “3D to structure data” sections in “How it Works.”)
Now, take a look at the MOL file in the text window. You will note that, as a default, Hack-a-Mol includes explicit H in the MOL files it generates. (See discussion of explicit and implicit H earlier in this module for more information.)
Identify the atoms and bonds that make up the ring. (These will vary depending on the way that you drew the molecule – the 2D sketch application numbers atoms and bonds in the order that they are drawn.) Remember, the first two columns in each bond table entry refer to rows in the atom table, and the third column gives the bond type (1=single, 2=double, etc.) connecting these two atoms. (You can check yourself by hovering over atoms in the 3D window or clicking the “labels” link above this window.)
Once you have identified the six ring bonds in the MOL file, manually adjust them to generate the other Kekulé structure of the ring. (That is, switch the 1’s for 2’s and the 2’s for 1’s in the bond type fields (third column) of the bond table entries for the six ring bonds.) With the cursor still in the text window, press enter. This should generate the other Kekulé structure for benzoic acid in both the 3D and 2D windows.
Just for kicks, let’s generate a nonsense structure. Change all of the ring bonds to double bonds, and press enter. You should now have a chemically-offensive structure involving a cyclohexahexene ring with six positively charged carbon atoms violating valence rules. There’s a lesson here – software won’t tell you that your structure data is chemically nonsensical unless it is programmed to do so.
Revert to benzoic acid, either by changing the bonds back manually or just by clearing the 2D sketch window, re-drawing, and clicking the right arrow button again.
Now, let’s stick a chlorine atom onto the benzene ring. Using the atom and bond tables, locate the atom table entry for a ring hydrogen ortho, meta, or para to the carboxyl group (your pick!). Change the atom symbol in this atom table entry from H to Cl, and press enter. You should now have the chlorobenzoic acid isomer of your choice in both 3D and 2D windows.
One more exercise: let’s make our benzoic acid into pyridine-3-carboxylic acid – that is, benzoic acid with N in place of one of the ring carbons meta to the carboxylic group. This is the compound better known as niacin (vitamin B3).
(Tangential fun fact: niacin, discovered as an acidic reaction product of nicotine, was originally named nicotinic acid. In the 1930s, it was found to be the essential nutrient that prevented pellagra, a devastating disorder widely prevalent in the American South in the early twentieth century. Public health officials promoted enriching flour with nicotinic acid, and the epidemic of pellagra began to disappear. However, physicians and scientists worried that the name “nicotinic acid” gave the impression that they were curing mass disease by putting tobacco into bread. A National Research Council committee decided to change the name of the substance to niacin, short for nicotinic acid vitamin.)
Anyway: locate the entry for a ring carbon meta to the carboxyl group. (Hint: 1) use the atom and bond tables to identify the carbon atom bonded to the two oxygen atoms; 2) find the ring carbon bonded to that carboxyl carbon; 3) find a ring carbon two bonds away from that carboxyl-substituted ring carbon.) Change that carbon to N, and press enter.
Now we have the N atom in our ring, but you will notice that it’s positively charged. We didn’t change any of the explicit hydrogens, so the N atom remains protonated, like the C atom that it replaced. Let’s get rid of that hydrogen atom. Locate the entry for the N-H bond in the bond table and the entry for the corresponding H atom in the atom table, and delete both of them. Press enter.
Unless you were very lucky, you should now have a monstrous mess in the 3D window and nothing at all in the 2D window. Uh-oh. Go back to the MOL file window, press ctrl-Z twice to undo the deletion of those rows, and press enter. That will take you back to N-protonated niacin.
By deleting a row of the atom table, we renumbered all of the subsequent atom table entries. Since we didn’t change the atom references in the bond table, this broke all of the bonds to these renumbered atoms.
Once again, delete that N-H bond from the bond table and the entry for that H atom in the atom table. However, now fix the bond table references by **decreasing the atom number by 1** for all atoms below the row that you deleted. (That is, if the hydrogen that you deleted was the 13th atom table entry, change each 14 in the first two columns of the bond table to a 13, and change each 15 in the first two columns of the bond table to a 14.)
Hit enter. Ugh – your structure is probably screwed up **again**, even if you did all of this renumbering correctly. You may even have lost your ring, for some reason.
Take a look at the counts line of the MOL file – the row above the atom table, just below the file headers. The first two numbers in this line refer to the number of atoms and bonds in the molecule. Since we deleted an atom and a bond, we need to decrease each of these from 15 to 14. Do so, and then press enter again. You should now have niacin.
Whew. Thank goodness that connection table handling is so amenable to automation!
Play around some more with Hack-a-Mol. Take a look at the “How it Works” page – a lot of the notations, apps, and processes referred to on this page will be covered in subsequent weeks. You may find it useful to continue to come back to this page and play around with it as you move on in this course.
EXERCISE
1. Does Hack-A-Mol handle the number 4 for an aromatic bond? How can you tell? Can you create a chemically sound but non-aromatic structure using 4s in the bond field?
2. Perfluorinated octanoic acid (PFOA) is a surfactant that played a key role for a long time in the manufacture of fluorinated polymers including Teflon. Over the past decade, it has been the subject of significant public health concern and a whole bunch of litigation.
Pull PFOA into Hack-a-Mol by typing it into the text search box below the 3D window and clicking “search.”
2a. Edit the mole file to defluorinate PFOA, converting it into octanoic acid.
2b. Now make it into acetic acid. (It is possible to do this in a way that yields correct-looking 2D and 3D renderings without changing any XYZ coordinately, but you have to be ***very*** careful about how you delete and relabel atoms and bonds.)
FURTHER READING
- https://en.wikipedia.org/wiki/Chemical_table_file
- CTFile Formats, June 2005, Elsevier/MDL, https://web.archive.org/web/20070630061308/http://www.mdl.com/downloads/public/ctfile/ctfile.pdf (Documentation for v2000 MOL file and related chemical table file formats.)
- Hack-a-Mol: https://chemapps.stolaf.edu/jmol/jsmol/hackamol.htm
- (Documentation: https://chemapps.stolaf.edu/jmol/docs/misc/hackamolworkings.pdf)