
2.4: Line Notation
- Page ID
- 154856
- Explain what SMILES, SMARTS and SMIRKS are.
- Explain what InChI and InChIKey are.
- Review SMILES specification rules.
- Compare and contrast SMILES and InChI.
- Demonstrate how to interpret SMILES, SMARTS, InChI strings into their corresponding chemical structures.
Introduction
Line notations represent structures as a linear string of characters. They are widely used in Cheminformatics because computers can easily process linear strings of data. Examples of line notations include the Wiswesser Line-Formula Notation (WLN)1, Sybyl Line Notation (SLN)2,3 and Representation of structure diagram arranged linearly (ROSDAL)4,5. Currently, the most widely used linear notations are the Simplified Molecular-Input Line-Entry System (SMILES)6-9 and the IUPAC Chemical Identifier (InChI)10-13, which are described below. In this class we will focus on SMILES and InChI line notation.
SMILES
The Simplified Molecular-Input Line-Entry System (SMILES)6-9 is a line notation for describing chemical structures using short ASCII strings. SMILES is like a connection table in that it identifies the nodes and edges of a molecular graph. SMILES was developed in the late 1980s and implemented by Daylight Chemical Information Systems (Santa Fe, NM), but it is still widely used today. A detailed information on SMILES can be found in Chapter 314 of the Daylight Theory Manual as well as the SMILES tutorial15.
SMILES Specification Rules
In SMILES, hydrogen are typically implicitly implied and atoms are represented by their atomic symbol enclosed in brackets unless they are elements of the “organic subset” (B, C, N, O, P, S, F, Cl, Br, and I), which do not require brackets unless they are charged. So gold would be [Au] but chlorine would be Cl. If hydrogens are explicitly implied brackets are used. A formal charge is represented by one of the symbols + or -. Single, double, triple, and aromatic bonds are represented by the symbols, -, =, #, and :, respectively. Single and aromatic bonds may be, and usually are, omitted. Here are some examples of SMILES strings.
| Function | Symbol | Function | Symbol | |
| single bond* | - | Positive charge | [C+] | |
| double bond | = | Negative charge | [C-] | |
| triple bond |
# |
aromatic carbon | c (lower case c) | |
| aromatic bond* | : |
Table 2.4.2 shows some common SMILES strings. Note the following conventions
- Branches are specified by enclosures in parentheses and can be nested or stacked, as shown in these examples.
- Rings are represented by breaking one single or aromatic bond in each ring, and designating this ring-closure point with a digit immediately following the atoms connected through the broken bond. Atoms in aromatic rings are specified by lower cases letters.
- Aromatic Rings use lower case c
- Although the carbon-carbon bonds in these two SMILES are omitted, it is possible to deduce that the omitted bonds are single bonds (for cyclohexane) and aromatic bonds (for benzene). One can also represent an aromatic compound as a non-aromatic, KeKulé structure. For example, the following is a valid SMILES string for benzene.
- C1=CC=CC=C1 Benzene (C6H6)
| SMILES | Name (formula) | SMILES | Name (formula) | SMILES | Name(formula) | ||
| C | Methane (CH4) | COC | Dimethyl ether (CH3OCH3) | CC(C)CO | Isobutyl alcohol (CH3-CH(CH3)-CH2-OH) | ||
| CC | Ethane (CH3CH3) | CCO | Ethanol (CH3CH2OH) | CC(CCC(=O)N)CN | 5-amino-4-methylpentanamide | ||
| C=C | Ethene (CH2CH2) | CC=O | Acetaldehyde (CH3-CH=O) | C1CCCCC1 | Cyclohexane (C6H12) | ||
| C#C | Ethyne (CHCH) | CC(=O)[O-] | Acetate | c1ccccc1 | Benzene (C6H6) (aromatic representation) |
||
| C#N | Hydrogen Cyanide (HCN) | [C-]#N | Cyanide anion | C1=CC=CC=C1 | Benzene (C6H6) (KeKulé representation) |
Note that aromaticity is not a measurable physical quantity, but a concept without a unanimous mathematical definition. As a result, different aromaticity detection algorithms often disagree with each other on whether a given molecule is aromatic or not, making it difficult to interchange information between databases that use different aromaticity detection algorithms for SMILES generation.
Also note that a ring structure can have multiple potential ring-closure points. For example, a six-membered ring has six bonds, each of which can be a ring-closure point. As a result, a ring compound may be represented by many different but equally valid SMILES strings. Actually, it is very common that there are a lot of SMILES strings that represent the same structure, whether it has a ring or not, because one can start with any atom in a molecule to derive a SMILES string. Therefore, it is necessary to select a “unique SMILES” for a molecule among many possibilities. Because this is done through a process called “canonicalization”, this unique SMILES string is also called the “canonical SMILES”.
Isomeric SMILES
Isomeric SMILES allow for the specification of the isotopism and stereochemistry of a molecule. Information on isotopism is indicated by the integral atomic mass preceding the atomic symbol. The atomic mass must be specified inside square brackets. For example, C-13 methane can be represented by “[13CH4]”. Configuration around double bonds is specified by “directional bonds” (characters / and \). For example, E- and Z-1,2-difluoroethene can be represented by the following isomeric SMILES:
- F/C=C/F or F\C=C\F (E)-1,2-difluoroethene (trans isomer)
- F/C=C\F or F\C=C/F (Z)-1,2-difluoroethene (cis isomer)
Configuration around tetrahedral centers are indicated by the symbols “@” or “@@”
- C[C@@H](C(=O)O)N L-Alanine
- C[C@H](C(=O)O)N D-Alanine
More detailed information on chirality specification can be found in Chapter 314 of the Daylight Theory Manual.
Limitations of SMILES
SMILES is proprietary and it is not an open project. This has led different chemical software developers to use different SMILES generation algorithms, resulting in different SMILES versions for the same compound. Therefore, SMILES strings obtained from different databases or research groups are not interchangeable unless they used the same software to generate the SMILES strings. With an aim to address this interchangeability issue of SMILES, an open-source project has launched to develop an open, standard version of the SMILES language called OpenSMILES. However, the most noticeable community effort in this area is development of InChI, which is described in next section.
SMARTS
SMiles ARbritrary Target Specification (SMARTS) notation allows one to search in certain databases (like PubChem) for generic structures. It is a language used for describing molecular patterns. SMARTS is useful for substructure searching, which finds a particular pattern (subgraph) in a molecule. SMARTS are straightforward extensions of SMILES. All SMILES symbols and properties are legal in SMARTS. SMARTS includes logical operators and additional molecular descriptors. Detailed information on SMARTS is given in the SMARTS specification document in the Daylight theory manual and SMARTS tutorial.
SMIRKS
Another extension of SMILES is SMIRKS, which is a line notation for generic reactions. A generic reaction represents a group of reactions that undergo the same set of atom and bond changes. Note that SMILES and SMARTS can be used to represent reactions, using the “>” symbol between the reactants, products, and agents, as described in the SMILES and SMARTS specification documents. (Therefore, these SMILES and SMARTS that describe reactions are often called reaction SMILES and reaction SMARTS, respectively.) On the other hand, SMIRKS is used to represent types of reactions (e.g., SN2 reaction). More detailed information on SMIRKS is given in the SMIRKS specification document and SMIRKS tutorial.
InChI
Since 1919 the International Union of Pure and Applied Chemistry (IUPAC) has been the international authority on chemical nomenclature and terminology. IUPAC currently consists of members from 57 national adhering organizations (NAOs) whose recommendations are made public through the IUPAC journal Pure and Applied Chemistry and the IUPAC Color books. As we entered the new millennial the leadership of IUPAC recognized the need to extend chemical nomenclature into the digital realm of computer databases and software agents, and in March of 2000 during a meeting at the U.S. Naval Academy started a project with the U.S. National Institute of Standards and Technology (NIST), to build a machine readable nomenclature standard, the InChI. It was originally called INChI for IUPAC/NIST Chemical Identifier, but was changed to InChI (International Chemical Identifier) as although it was built with efforts from NIST, it was not appropriate for NIST as a government agency to place its name as a recommendation for the identifier. In 2010 the InChI Trust was formed and development of the standard is continuing the purview of the InChI Trust and the IUPAC InChI subcommittee.
InChI is an open, freely available non-proprietary computer generated chemical identifier that is based on a hierarchical layered line notation (see below). The first three layers essentially deal with the information within the simplified connection table, and the additional layers are added as needed, and deal with complexities like isomers, isotopic distributions and the other types of issues brought up in section 2.3 of this chapter, and these layers are extensible. A standard InChI has a predefined number of layers, and these can be extended to non-standard InChI's that can have new layers relating to define additional information, that is what is meant by extensible layes. Unlike SMILES, InChI is a canonical line notation and so is a unique identifier that is built upon a set of nomenclature rules. That is, although there are canonical SMILES built through a canonicalization algorithm, there can be more than one canonicalization algorithm for SMILES, and so you can have more than one SMILES string for the same structure.
Students may be familiar with the American Chemical Society's Chemical Abstract Service (CAS) registry number, which is supposed to be a unique identifier based on the registry system, but issues can arise (see below, other identifiers, and problem). Also, a CAS registry number is associated with a compound that has been published in the primary literature or patents, and the CAS system bases its identifiers on the registry system, not the structure of the molecule. That is, InChI is not a registry system, it is a type of nomenclature that describes the structure of a molecule, and you can make an InChI for a molecule that does not exist, as long as you specify its structure.
The most recent version of InChI (and its documentation) can be obtained at the InChI Trust Download site.
InChI: A Layered Notation
The power of a layered notation is that it gets to the essence of what is a molecule? For example, we think water is H2O, but if you look at a real sample of water you will note that some of the hydrogens are protium (one proton) and others are deuterium ( a proton and a neutron), and in fact the ratio of deuterium to protium in ground water samples can vary from one region of the US to another, and thus the molar mass of samples of water can vary. In fact IUPAC has now adopted an "interval atomic weight notation system" for some elements whose atomic mass varies across samples, and this can affect physical properties of a sample, like the vapor pressure of water. So water is water, but not all water is the same, and the question becomes, do you care? If you are uploading data to a database and know the isotopic distribution you care, but if you do not know it, you do not care, but in both cases, your data deals with water. Through a layered notation system you can have an isotopic layer to describe your water if you care, but you don't need it if you don't care. This leads to one of the issues that comes up with the layered notation, in that you can have different InChI's for a compound, depending on the kind of information you want in the name, that is how many and what kind of layers you use. This leads to the standard InChI, which is an InChI that has defined layers and is thus canonical, and starts with a number followed by the letter "s" to indicate the version of the standard.
Standard InChI
Standard InChI version 1.05 was released in January 2017 and has 6 core layers (and several sublayers within the core layers) and starts with InChI=1S/... . Each layer in the InChI string is separated by a "/" and the "main layer" is essentially the connection table. The InChI software generates both standard and nonstandard InChI, with the standard InChI having "fixed options" that ensures interoperability between databases and software agents. The standard InChI (version 1.05) has the following layers:
- Main Layer
- Chemical Formula Layer (based on Hill Notation)
- Connections- bonds between atoms and may have sublayers, with the last one dealing with mobile hydrogens.
- Charge Layer
- Component Charge
- Protons
- Stereochemical Layer
- Double Bond sp2 (Z/E) Sterochemistry
- Tetrahedral Sterochemistry
- Isotopic Layer
- Fixed Hydrogen Layer (binds mobile hydrogens)
- Polymer Layer (actually a new experimental layer) and does not affect the content of the earlier layers.
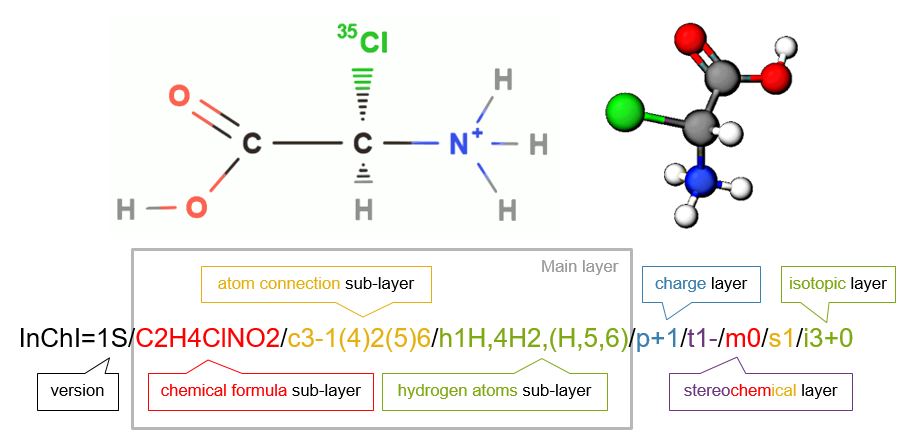
Figure \(\PageIndex{1}\): The main layers for a standard InChI of [(R)-carboxy(chloro)methyl]azanium, the protonated form of 2-(35Cl)chloro-R-glycine . Note each layer or sublayer is separated by a forward slash [/].
NonStandard InChI
Note a nonstandard InChI does not start with InChI=1S/... but with a InChI=1/.... and has additional layers that approach different facets of a molecule's structure or features. In fact a company could create their own lawyer for a nonstandard information and encode into it proprietary information that they wished to keep private. The nonstandard InChI may not be canonical, but can handle facets of information information that a standard can not, in fact for a standard to be canonical different tautomers must have the same InChI, or you have two InChIs for the same molecule
So defining specific tautomers is one use of a nonstandard InChI as can be seen in the case of 4,5-Dihydro-1,3-Oxazol-3-lum. Figure 2 shows the two tautomeric forms of this molecule which must have the same standard InChI or it would not be canonical (you would have two InChIs for the same molecules). If you want to define just one of the tautomers, you need to use a nonstandard InChI and add a fixed hydrogen layer (in red). Although these are two ways of drawing the same molecule, one form may be favored over the other in certain environments and so there may be data indicative of the behavior of one of these form and not the other, and thus there may be a need to distinguish between the tautomers.
| InChI=1S/C3H5NO/c1-2-5-3-4-1/h3H,1-2H2/p+1 | |
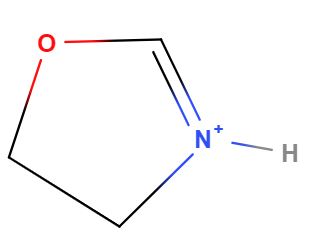 |
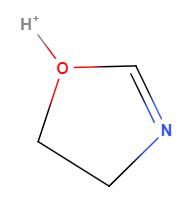 |
| InChI=1/C3H5NO/c1-2-5-3-4-1/h3H,1-2H2/p+1/fC3H6NO/h4H/q+1 | InChI=1/C3H5NO/c1-2-5-3-4-1/h3H,1-2H2/p+1/fC3H6NO/h5H/q+1 |
Figure \(\PageIndex{2}\): The above two structures are tautomeric drawings of the same molecule and thus have the same standard InChI. If you were interested in just one of the structures you could use a nonstandard InChI with a fixed hydrogen layer (in red). Borrowed from section 6.2 of InChI Trust FAQ.
Drawbacks of InChI
InChIs are not meant to be human readable but to contain molecular information that computers can read within the layers, so unlike SMILES you can't really read even a simple InChI (see Figure 3), never mind a complex one (figure 4).
| SMILES | InChI |
|---|---|
| CC(=O)C | InChI=1S/C3H6O/c1-3(2)4/h1-2H3 |
|
Acetone |
|

Figure \(\PageIndex{3}\): Canonical SMILES and InChI for Acetone (source: PubChem)
Another drawback of InChI is just like an IUPAC systematic name, they are of variable length and become real long (figure 4). The problem with the variable length is it makes InChI impractical as a database registry number, and the length is often too long for internet search engines to handle.
| IUPAC Systematic Name | InChI |
|---|---|
| magnesium;methyl (3R,21S,22S)-16-ethenyl-11-ethyl-12,17,21,26-tetramethyl-4-oxo-22-[3-oxo-3-[(E,7R,11R)-3,7,11,15-tetramethylhexadec-2-enoxy]propyl]-23,25-diaza-7,24-diazanidahexacyclo[18.2.1.15,8.110,13.115,18.02,6]hexacosa-1,5,8(26),9,11,13(25),14,16,18,20(23)-decaene-3-carboxylate | InChI=1S/C55H73N4O5.Mg/c1-13-39-35(8)42-28-44-37(10)41(24-25-48(60)64-27-26-34(7)23-17-22-33(6)21-16-20-32(5)19-15-18-31(3)4)52(58-44)50-51(55(62)63-12)54(61)49-38(11)45(59-53(49)50)30-47-40(14-2)36(9)43(57-47)29-46(39)56-42;/h13,26,28-33,37,41,51H,1,14-25,27H2,2-12H3,(H-,56,57,58,59,61);/q-1;+2/p-1/b34-26+;/t32-,33-,37+,41+,51-;/m1./s1 |
|
Chlorophyll A |
|
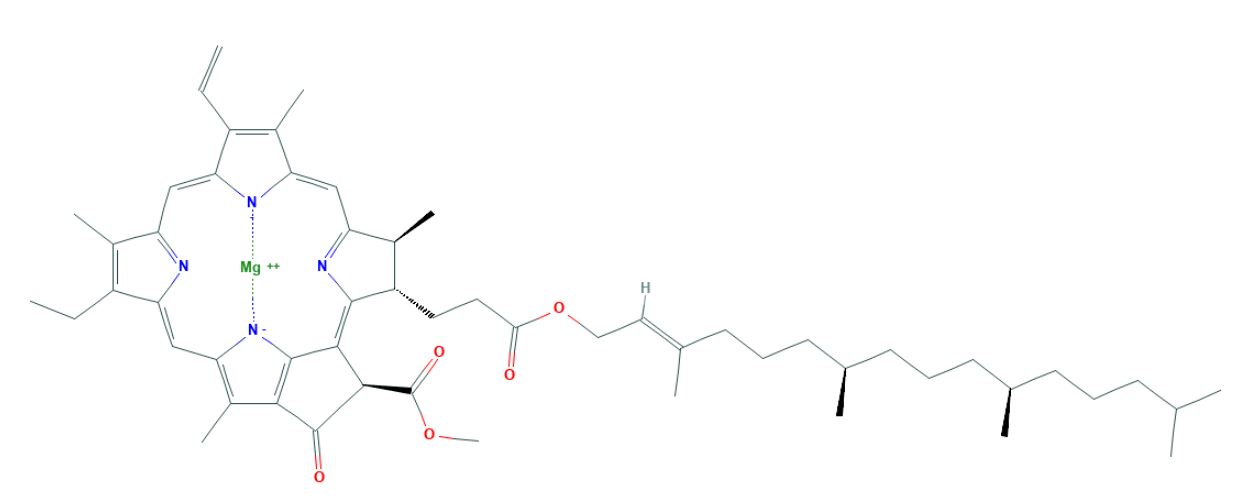
Figure \(\PageIndex{4}\): On the top left is the IUPAC systematic name for chlorophyll A and on the right is it's InChI (source: PubChem).
There is an additional issue with the InChI in that some of the characters interfere with web search queries and thus the InChI itself is not appropriate for web searches. To solve these problems a hashed InChI Key has been developed which is of constant length and enables web searches. The hashed key is also of constant length, making it better suited for databases.
InChI Keys
The InChI suite will generate a hashed version of the InChI, the InChI Key. The hash function generates a standard key of 27 characters that stores information in four parts (see figure 5). The InChIKey may be a standard or nonstandard key as indicated by the version, but all keys are of the same length and format.

Figure \(\PageIndex{5}\): InChI key for 2-(35Cl)chloro-R-glycine (molecule in figure 1).
The hash function is a one-way conversion (figure 6), that is, if you have an InChI you can generate the key, but if you have the key you can not generate the InChI. The key can function as an identifier is you made it registry number where you would need a look up table to know the molecule it is associated with.

Figure \(\PageIndex{6}\): The one way InChIKey generation function.
If two different chemical compound databases have the same chemical (InChI)they will generate the same standard InChIKeys and thus it is customary for databases and other information sources like Wikipedia chemboxes to generate standard InChIKeys, and they effectively function as a standard "registry number", that is, if two chemicals in different databases have the same standard InChIKey, they are the same chemical. On the other hand if you had a non-standard InChI the non-standard layers would induce variability of the key and so you could not compare across databases.
InChIKeys and Web Searching
The molecule (R)-2-(35Cl)chloroglycine probably does not exist and was created to demonstrate the layers of an InChI and the correlating key. If you do a web search of the entire key (UWPWWENWLZPQGU-WRFRXMDISA-0) you do not get any hits, but if you search just the main layer you get several hits. What you are doing is essentially looking for any molecule with the same simplified connection table, that is, all stereoisomers, or isotopic labels. One of the hits is for [(S)-carboxy(chloro)methyl]azanium which is the other isomer. If you go to properties they are all computed and none were deposited to PubChem by vendors or contributors, and so this molecule has probably never been synthesized. Under 5.2 Related Compounds/Exact Same Parent you also get the non-protonated form (2-chloro-L-glycine) , of which there is published information. It is also of interest that the search of first part of the InChIKey also turned up an article in Russian on the L isomer, (you may need to download the pdf to see the actual bonds).
InChI OER
The InChI Trust runs an Open Education Resource (OER) where you can find material on InChI https://www.inchi-trust.org/oer/. The InChI OER is a repository where anyone can upload and tag material on InChI, or link to and tag existing material on the use of InChI. Once material is posted within the OER it can be searched through a filter system.
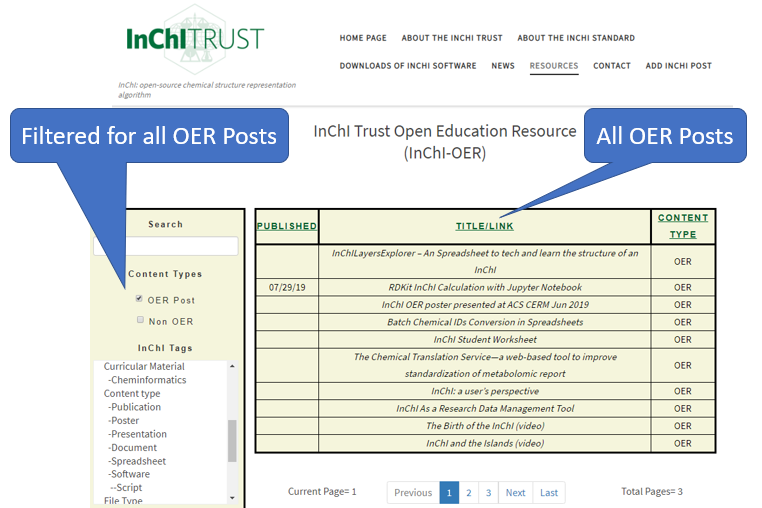
Figure \(\PageIndex{7}\): InChI OER tag filter and associated content. The default setting is to show all OER site material, clicking non-OER will extend the filter to include off site material like publications which have records that have been submitted to the OER.
InChI Layers Explorer
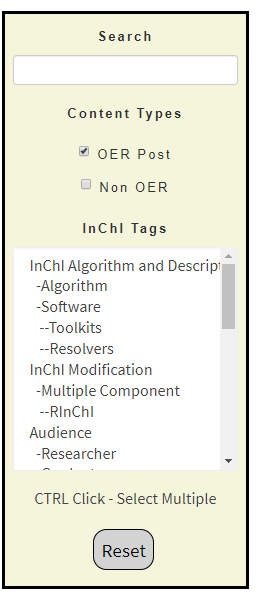
In this activity we will use the InChI OER to obtain an Excel spreadsheet that breaks an InChI into layers, and start to analyze how cheminformatics functionality can be integrated into common tools like spreadsheets. Go to the InChI OER and in the filter click "Spreadsheet" (middle of figure \(\PageIndex{8}\)). This filters the content to items that are tagged "spreadsheet" and also removes any tag that is not associated with one of those content items. Now move down to tag category "File Type" and while holding the <ctrl> key, click Excel (right figure \(\PageIndex{8}\)). You now get a list of excel spreadsheets (figure \(\PageIndex{9}\)).
 |
 |
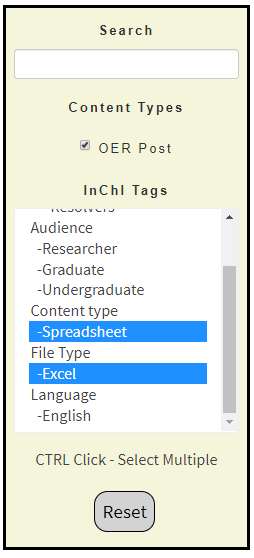 |
Figure \(\PageIndex{8}\): InChI OER Tag Filter.
On the left is the default setting and all content loaded to the site is displayed in the window (right side of figure \(\PageIndex{7}\). In the middle the filter for spreadsheets is activated, and you can see there are two types that have been uploaded, Google Sheets and Excel sheets. On the right both Spreadsheet and Excel have been activated, and so only spreadsheets in Excel are displayed and the content view is reduced to those items that are tagged both "Spreadsheet" and "Excel" (Figure \(\PageIndex{9}\))

Figure \(\PageIndex{9}\): At the time this page was created there were three items uploaded to the InChI OER that were tagged as Excel Spreadsheets.
Click on the InChILayersExplorer and you go to it's content page. This page will have a description of the content and a green information box (Figure \(\PageIndex{10}\)), and in the information box is a "Download Publication Files", that allows you to obtain the spreadsheet.
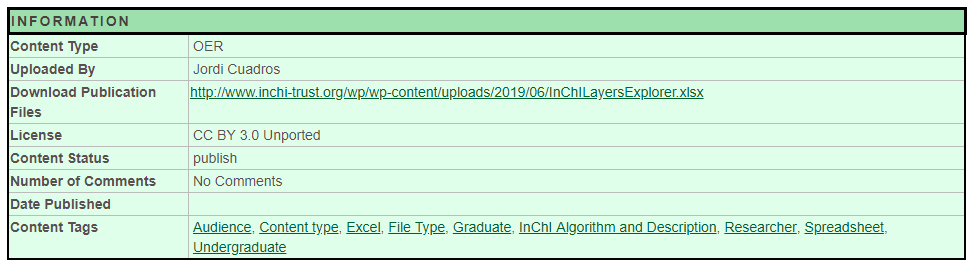
Figure \(\PageIndex{10}\): Green Information box for the InChILayersExplorer
Now click on the link in the "Download Publications File" field and you will have a copy of the InChI Layers Explorer, which you should open and enable editing.
Using the InChILayersExplorer show the difference between the InChI for (R)-thalidomide and (S)-thalidomide. Note, the goal of this activity is not to answer the question, but to gain an understanding on how the InChILayersExplorer works, which is in effect a "smart spreadsheet" that communicates with database web APIs via webservices functions. One of the skills we hope you can gain from this class is enough familiarity with how code works so if you see new code, you can hack in and figure how it works. Be sure to enable the spreadsheet after you download it. This spreadsheet communicates with the NCI Chemical Resolver (section 2.7. )
1. Type (R)-thalidomide in the yellow region (type over CoA), OK, it fails, now try the (S) isomer, and it still fails, so now try thalidomide without specifying an isomer. OK, so you have the InChI for thalidomide, but there is nothing in the stereochemical layer, as you have not specified the stereochemistry. These spreadsheet uses the Chemical Identifier Resolver of the NIH which will be covered in section 2.6.2.1.1), which can be accessed directly at https://cactus.nci.nih.gov/chemical/structure and is shwon in figure \(\PageIndex{11}\). Now lets start by searching for (R)-thalidomide directly in the resolver (figure \(\PageIndex{11}\)).
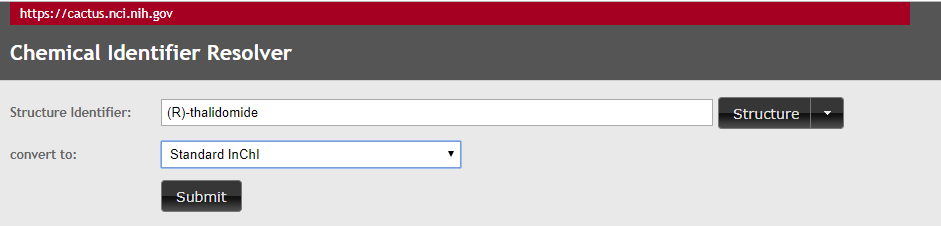
Figure \(\PageIndex{11}\): NCI/CADD Chemical Resolver set up to find standard InChI for (R)-thalidomid
As you may have guessed, neither (R) or (S) works, but "thalidomide" does (incidentally, you have to hit submit, not Structure), and so this resolver will not provide information on the isomers of thalidomide. So now do a web search of (R)-thalidomide, and paste in its key (UEJJHQNACJXSKW-SECBINFHSA-N), and note the stereochemical layer [/t9-/m1/s1] is the only part that is different. Now repeating for (S)-thalidomide.
You should get the following results
Table \(\PageIndex{3}\)
| Compound | InChI Key | Stereochemical layer |
| thalidomide | UEJJHQNACJXSKW-UHFFFAOYSA-N | none |
| (R)-thalidomide | UEJJHQNACJXSKW-SECBINFHSA-N | /t9-/m1/s1 |
| (S)-thalidomide | UEJJHQNACJXSKW-VIFPVBQESA-N | /t9-/m0/s1 |
Note, if you click on the merged cells that generates the InChI (Rows 7-8) you see the following code.

Figure \(\PageIndex{11}\): Code in spreadsheet that uses WEBSERVICE function to get InChI from NCI/CADD chemical resolver
Now open up a browser tab and paste in the following URL:
https://cactus.nci.nih.gov/chemical/structure/thalidomide/stdinchi
Now go back to the NCI Chemical Resolver and click the dropdown box of the "convert to" field (figure\(\PageIndex{12}\) and try another option, say "TwirlyMol(3D).
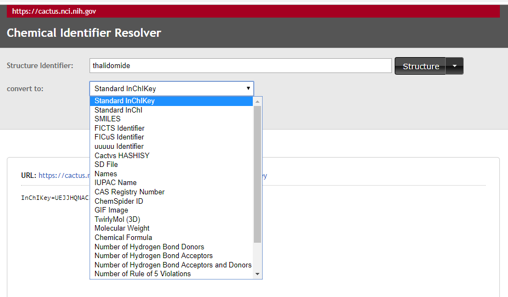
\(\PageIndex{12}\): Dropdown menu of NCI Chemical resolver showing some of the options.
Can you figure out the URL that uses the NCI Chemical Resolver to give the 3D molecule in a webpage? Once you have done this, can you identify a problem that has resulted from these molecular representations. Hint, think of adding two more columns to table \(\PageIndex{3}\), one for 2D and one for 3D images. What is the issue when you draw the 3D image that does not arise when you draw the 2D?
References and Further Reading
- (1) Wiswesser, W. J. J. Chem. Inf. Comput. Sci. 1982, 22, 88.
- (2) Ash, S.; Cline, M. A.; Homer, R. W.; Hurst, T.; Smith, G. B. J. Chem. Inf. Comput. Sci.1997, 37, 71.
- (3) Homer, R. W.; Swanson, J.; Jilek, R. J.; Hurst, T.; Clark, R. D. J. Chem Inf. Model.2008, 48, 2294.
- (4) Barnard, J. M.; Jochum, C. J.; Welford, S. M. Acs Symposium Series 1989, 400, 76.
- (5) Rohbeck, H. G. In Software Development in Chemistry 5; Gmehling, J., Ed.; Springer Berlin Heidelberg: 1991, p 49.
- (6) Weininger, D. J. Chem. Inf. Comput. Sci. 1988, 28, 31.
- (7) Weininger, D.; Weininger, A.; Weininger, J. L. J. Chem. Inf. Comput. Sci. 1989, 29, 97.
- (8) Weininger, D. J. Chem. Inf. Comput. Sci. 1990, 30, 237.
- (9) SMILES: Simplified Molecular Input Line Entry System (http://www.daylight.com/smiles/) (Accessed on 6/30/2015).
- (10) Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. J. Cheminform. 2013, 5, 7.
- (11) Heller, S.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. J. Cheminform. 2015, 7, 23.
- (12) The IUPAC International Chemical Identifier (InChI) (http://www.iupac.org/home/publications/e-resources/inchi.html) (Accessed on 6/29/2015).
- (13) InChI Trust (http://www.inchi-trust.org/) (Accessed on 6/29/2015).
- (14) Daylight Theory Manual, Chapter 3: SMILES - A Simplified Chemical Language (http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html) (Accessed on 6/23/2015).
- (15) Daylight SMILES Tutorial (http://www.daylight.com/dayhtml_tutorials/languages/smiles/index.html) (Accessed on 6/23/2015).
Contributors
Robert E. Belford (University of Arkansas Little Rock; Department of Chemistry). The breadth, depth and veracity of this work is the responsibility of Robert E. Belford, rebelford@ualr.edu. You should contact him if you have any concerns. This material has both original contributions, and content built upon prior contributions of the LibreTexts Community and other resources, including but not limited to:
- Sunghwan Kim
- Material Adapted from 2017 Cheminformatics OLCC