6.2.5: Metal Phosphines
- Page ID
- 360907
Learning Objectives
In this lecture you will learn the following
- Know about metal phosphine complexes.
- Have an understanding of the steric and electronic properties of the phosphine ligands.
- Obtain a deeper insight about the metal phosphine interactions.
- Be introduced to other π−basic ligands.
Phosphines are one of the few ligands that have been extensively studied over the last few decades to an extent that the systematic fine tuning of the sterics and electronics can now be achieved with certain degree of predictability. Phosphines are better spectator ligands than actor ligands. Tolman carried out pioneering infrared spectroscopy experiments on the PR3Ni(CO)3 complexes looking at the ν(CO)stretching frequencies for obtaining an insight on the donor properties of the PR3 ligands. Thus, a stronger σ−donor phosphine ligand would increase the electron density at the metal center leading to an enhanced metal to ligand π−back bonding and thereby lowering of the ν(CO) stretching frequencies in these complexes. Another important aspect of the phosphine ligand is its size that has significant steric impact on its metal complexes. Thus, unlike CO ligand, which is small and hence many may simultaneously be able to bind to a metal center, the same is not true for the phosphine ligands as only a few can bind to a metal center. The number of phosphine ligands that can bind to a metal center also depends on the size of its R substituents. For example, up to two can bind to a metal center in case of the PCy3 or P(i−Pr)3 ligands, three or four for PPh3, four for Me2PH, and five or six for PMe3. The steric effect of phosphine was quantified by Tolmann and is given by a parameter called Cone Angle that measures the angle at the metal formed by the PR3 ligand binding to a metal (Figure \(\PageIndex{1}\)).
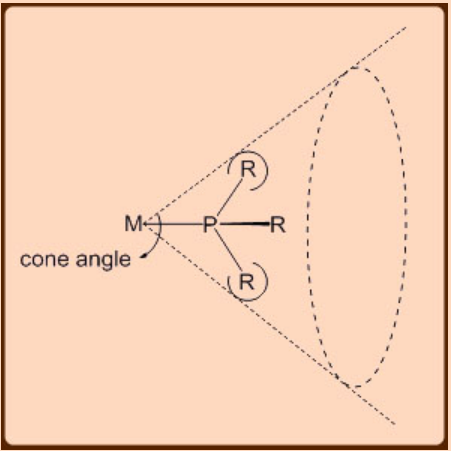
The Cone Angle criteria has been successfully invoked in rationalizing the properties of a wide range of metal phosphine complexes. One unique feature of the phosphine ligand is that it allows convenient change of electronic effect without undergoing much change in its steric effects. For example, PBu3 and P(OiPr)3 have similar steric effects but vary in their electronic effects. The converse is also true as the steric effect can be easily changed without undergoing much change in the electronic effect. For example, PMe3 and P(o−tolyl)3 have similar electronic effect but differ in their steric effects. Thus, the ability to conveniently modulate the steric and the electronic effects make the phosphine ligands a versatile system for carrying out many organometallic catalysis.
Structure and Bonding
Phosphines are two electron donors that engage a lone pair for binding to metals. These are thus considered as good σ−donors and poor π−acceptors and they belong to the same class with the aryl, dialkylamino and alkoxo ligands. In fact they are more π−acidic than pure σ−donor ligands like NH3and, more interestingly so, their π−acidity can be varied significantly by systematic incorporation of substituents on the P atom. For example, PF3 is more π−acidic than CO. Analogous to what is observed in case of the benchmark π−acidic CO ligand, in which the metal dπ orbital donates electron to a π* orbital of a C−O bond, in the case of the phosphines ligands, such π−back donation occurs from the metal dπ orbital occurs on to a σ* orbital of a P−R bond (Figure \(\PageIndex{2}\)). In phosphine ligands, with the increase of the electronegativity of R both of the σ and the σ* orbitals of the P−R bond gets stabilized. Consequently, the contribution of the atomic orbital of the P atom to the σ*−orbital of the P−R bond increases, which eventually increases the size of the σ* orbital of the P−R bond. This in turn facilitates better overlap of the σ* orbital of the P−R bond with the metal dπ orbital during the metal to ligand π−back donation in these metal phosphine complexes.
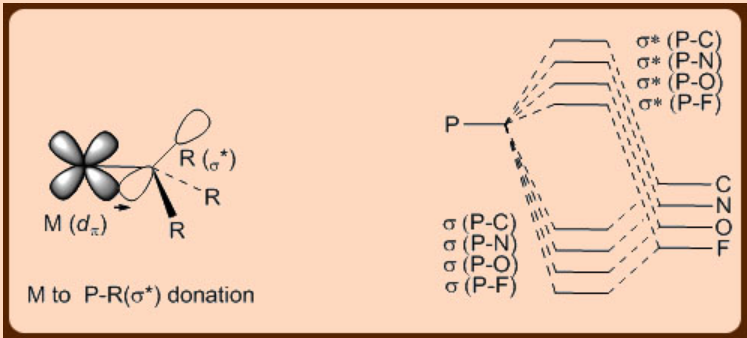
Starting from CO, which is a strong π−acceptor ligand, to moving to the phosphines, which are good σ−donors and poor π−acceptor ligands, to even going further to other extreme to the ligands, which are both good σ−donors as well as π−donors, a rich variety of phosphine ligands thus are available for stabilizing different types of organometallic complexes. In this context the following ligands are discussed below.
π-basic ligands
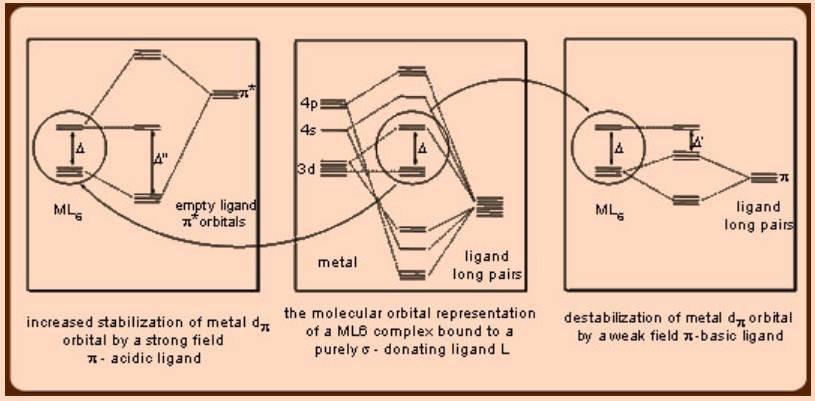
Alkoxides (RO−) and halides like F−, Cl− and Br− belong to a category of π−basic ligands as they engage a second lone pair for π−donation to the metal over and above the first lone pair partaking σ−donation to the metal. Opposite to what is observed in the case of π−acidic ligands, in which the π* ligand orbital stabilizes the dπ metal orbital and thereby affecting a larger ligand field splitting, as consistent with the strong field nature of these ligands (Figure 3), in the case of the π−basic ligands, the second lone pair destabilizes the dπ metal orbitals leading to a smaller ligand field splitting, which is in agreement with the weak field nature of these ligands. The orbitals containing the lone pair of the ligands are usually located on the more electronegative heteroatoms and so they are invariably lower in energy than the metal dπ orbitals. Hence, the destabilization of the metal dπ orbitals occurs due to the repulsion of the filled ligand lone pair orbital with the filled metal dπ orbitals. In case of the situations in which the metal dπ orbitals are vacant, like in d0 systems of Ti4+ ions, the possibility of the destabilization of the metal dπ orbitals do not arise but instead stabilization occurs through the donation of the filled ligand lone pair orbital electrons to the empty metal dπ orbitals as seen in the case of TiF6 and W(OMe)6. Thus, this scenario in π−basic ligands is opposite to that observed in case of the π−acidic ligands, for which the empty π* ligand orbitals are higher in energy than the filled metal dπ orbitals.