4.5: Diatomic Molecules
- Page ID
- 344438
Valence bond theory fails for a number of the second row diatomics, most famously for O2, where it predicts a diamagnetic, doubly bonded molecule with four lone pairs. O2 does have a double bond, but it has two unpaired electrons in the ground state, a property that can be explained by the MO picture. We can construct the MO energy level diagrams for these molecules as follows
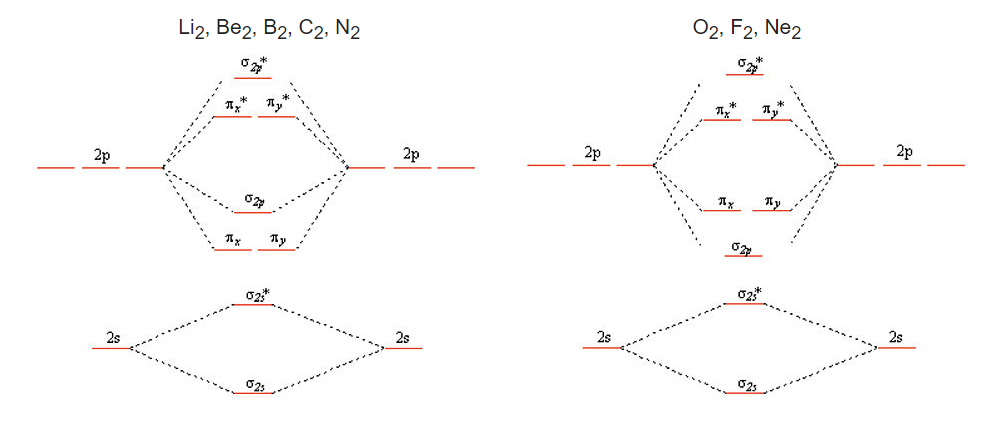
We get the simpler diatomic MO picture on the right when the 2s and 2p AOs are well separated in energy, as they are for O, F, and Ne. The picture on the left results from mixing of the σ2s and σ2p MO’s, which are close in energy for Li2, Be2, B2, C2, and N2. The effect of this mixing is to push the σ2s* down in energy and the σ2pup, to the point where the pπ orbitals are below the σ2p. Asymmetric diatomic molecules and ions such as CO, NO, and NO+ also have the ordering of energy levels shown on the left because of sp mixing. A good animation of the molecular orbitals in the CO molecule can be found on the University of Liverpool Structure and Bonding website.
Why don't we get sp-orbital mixing for O2 and F2? The reason has to do with the energies of the orbitals, which are not drawn to scale in the simple picture above. As we move across the second row of the periodic table from Li to F, we are progressively adding protons to the nucleus. The 2s orbital, which has finite amplitude at the nucleus, "feels" the increased nuclear charge more than the 2p orbital. This means that as we progress across the periodic table (and also, as we will see later, when we move down the periodic table), the energy difference between the s and p orbitals increases. As the 2s and 2p energies become farther apart in energy, there is less interaction between the orbitals (i.e., less mixing).
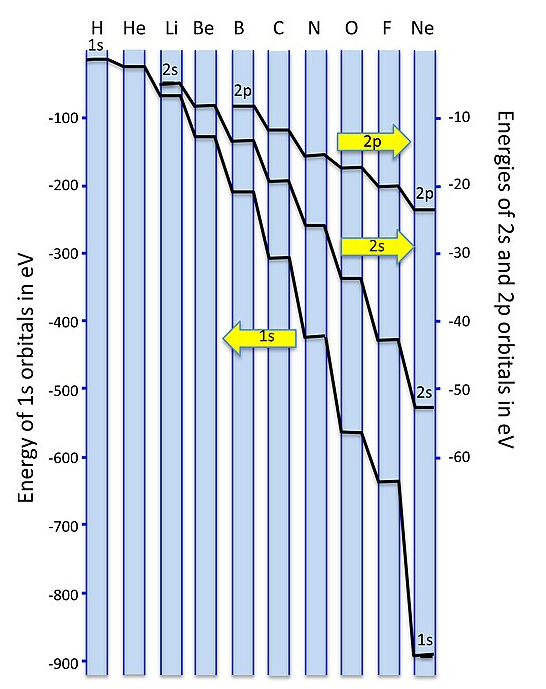
A plot of orbital energies is shown below. Because of the very large energy difference between the 1s and 2s/2p orbitals, we plot them on different energy scales, with the 1s to the left and the 2s/2p to the right. For elements at the left side of the 2nd period (Li, Be, B) the 2s and 2p energies are only a few eV apart. The energy difference becomes very large - more than 20 electron volts - for O and F. Since single bond energies are typically about 3-4 eV, this energy difference would be very large on the scale of our MO diagrams. For all the elements in the 2nd row of the periodic table, the 1s (core) orbitals are very low in energy compared to the 2s/2p (valence) orbitals, so we don't need to consider them in drawing our MO diagrams.