
9.2B: Solving the H₂⁺ System Exactly (Optional)
- Page ID
- 198719
The hydrogen molecule ion \(H_{2}^{+}\) is the only molecule for which we can solve the electronic Schrödinger equation exactly (and only within the Born Oppenheimer Approximation). Note that it has just one electron! In fact, there are no multi-electron molecules we can solve exactly. Thus from \(H_2\) on up to more complicated molecules, we only have approximate solutions for the allowed electronic energies and wavefunctions. Before we discuss these, however, let us examine the exact solutions for \(H_{2}^{+}\) starting with a brief outline of how the exact solution is carried out.
Figure \(\PageIndex{1}\) shows the geometry of the \(H_{2}^{+}\) molecule ion and the coordinate system we will use.
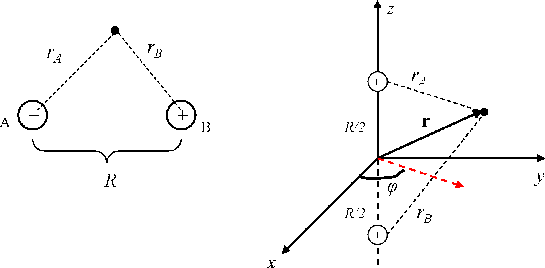
The two protons are labeled A and B, and the distances from each proton to the one electron are \(r_A\) and \(r_B\), respectively. Let \(R\) be the distance between the two protons (this is the only nuclear degree of freedom that is important, and the electronic wavefunction will depend parametrically only on \(R\)). The coordinate system is chosen so that the protons lie on the z-axis one at a distance \(R/2\) above the \(xy\) plane and one a distance \(-R/2\) below the \(xy\) plane. The classical energy of the electron is
\[\frac{p^2}{2m_e}-\frac{e^2}{4\pi \epsilon_0}\left ( \frac{1}{r_A}+\frac{1}{r_B} \right )=E\]
The nuclear-nuclear term \(V_{nn}(R)=e^2 /4\pi \epsilon_0 R\) is a constant, and we can define the potential energy relative to this quantity.
The energy is not a simple function of energies for the \(x\), \(y\), and \(z\) directions, so we try another coordinate system to see if we can simplify the problem. In fact, this problem has a natural cylindrical symmetry (analogous to the spherical symmetry of the hydrogen atom) about the z-axis. Thus, we try cylindrical coordinates. In cylindrical coordinates the distance of the electron from the z-axis is denoted \(\rho\), the angle \(\phi\) is the azimuthal angle, as in spherical coordinates, and the last coordinate is just the Cartesian \(z\) coordinate. Thus,
- \(x=\rho \cos\phi\)
- \(y=\rho \sin\phi \)
- \(z=z\)
Using right triangles, the distance \(r_A\) and \(r_B\) can be shown to be
\[r_A =\sqrt{\rho^2 +(R/2-z)^2}\;\;\;\; r_B =\sqrt{\rho^2 +(R/2 +z)^2}\]
The classical energy becomes
\[\frac{p_{\rho}^{2}}{2m_e}+\frac{p_{\phi}^{2}}{2m_e \rho^2}+\frac{p_{z}^{2}}{2m_e}-\frac{e^2}{4\pi \epsilon_0}\left ( \frac{1}{\sqrt{\rho^2 +(R/2-z)^2}}+\frac{1}{\sqrt{\rho^2 +(R/2+z)^2}} \right ) =E\]
First, we note that the potential energy does not depend on \(\phi\), and the classical energy can be written as a sum \(E=\varepsilon_{\rho ,z}+\varepsilon_\phi\) of a \(\rho\) and \(z\) dependent term and an angular term. Moreover, angular momentum is conserved as it is in the hydrogen atom. However, in this case, only one component of the angular momentum is actually conserved, and this is the z-component, since the motion is completely symmetric about the z-axis. Thus, we have only certain allowed values of \(L_z\), which are \(m\bar{h}\), as in the hydrogen atom, where \(m=0,\pm 1,\pm 2,...\). The electronic wavefunction (now dropping the "(elec)" label, since it is understood that we are discussing only the electronic wavefunction), can be written as a product
\[\psi (\rho ,\phi ,z)=G(\rho ,z)y(\phi)\]
and \(y(\phi )\) is given by
\[y_m (\phi )=\frac{1}{\sqrt{2\pi}}e^{im\phi}\]
which satisfies the required boundary condition \(y_m (0)=y_m (2\pi )\). Unfortunately, what is left in \(\rho\) and \(z\) is still not that simple. But if we make one more change of coordinates, the problem simplifies. We introduce two new coordinates \(\mu\) and \(\nu\) defined by
\[\mu =\frac{r_A +r_B}{R}\;\;\;\; \nu =\frac{r_A -r_B}{R}\]
Note that when \(\nu =0\), the electron is in the \(xy\) plane. Thus, \(\nu \) is analogous to \(z\) in that it varies most as the electron moves along the z-axis. The presence or absence of a node in the \(xy\) plane will be an important indicator of wavefunctions that lead to a chemical bond in the molecule or not. The coordinate \(\mu \), on the other hand, is minimum when the electron is on the z-axis and grows as the distance of the electron from the z-axis increases. Thus, \(\mu \) is analogous to \(\rho\). The advantage of these coordinates is that the wavefunction turns out to be a simple product
\[\psi (\mu ,\nu ,\phi ,R)=M(\mu )N(\nu )y(\phi )\]
which greatly simplifies the problem and allows the exact solution.
The mathematical structure of the exact solutions is complex and nontransparent, so we will only look at these graphically, where we can gain considerably insight. First, we note that the quantum number \(m\) largely determines how the solutions appear. First, let us introduce the nomenclature for designating the orbitals (solutions of the Schrödinger equation, wavefunctions) of the system
- If \(m=0\), the orbitals are called \(\sigma\) orbitals, analogous to the \(s\) orbitals in hydrogen.
- If \(m=1\), the orbitals are called \(\pi\) orbitals, analogous to the \(p\) orbitals in hydrogen.
- If \(m=2\), the orbitals are called \(\delta\) orbitals, analogous to the \(d\) orbitals in hydrogen.
- If \(m=3\), the orbitals are called \(\phi\) orbitals, analogous to the \(f\) orbitals in hydrogen.
These orbitals are known as molecular orbitals because the describe the electronic wavefunctions of an entire molecule. There are four designators that we use to express each molecular orbital:
- A greek letter, \(\sigma\), \(\pi\), \(\delta\), \(\phi\), .... depending on the quantum number \(m\).
- A subscript qualifier g or u depending on how an orbital \(\psi\) behaves with respect to a spatial reflection or parity operation \(r\rightarrow -r\). If \[\psi (-r)=\psi (r)\] then \(\psi (r)\) is an even function of \(r\), so we use the \("g"\) designator, where \(g\) stands for the German word gerade, meaning "even". If \[\psi (-r)=-\psi (r)\] then \(\psi (r)\) is an odd function of \(r\), and we use the \("u"\) designator, where \(u\) stands for the German word ungerade, meaning "odd".
- An integer \(n\) in front of the Greek letter to designate the energy level. This is analogous to the integer we use in atomic orbitals \((1s, 2s, 2p,...)\).
- An asterisk or no asterisk depending on the presence or absence of nodes between the nuclei. If there is significant amplitude between the nuclei, then the orbital favors a chemical bond, and the orbital is called a bonding orbital. If there is a node between the nuclei, the orbital does not favor bonding, and the orbital is called an antibonding orbital.
So, the first few orbitals, in order of increasing energy are:
\[1\sigma_g ,1\sigma_{u}^{*},2\sigma_g ,2\sigma_{u}^{*}, 1\pi_u ,3\sigma_g ,1\pi_{g}^{*},3\sigma_u\]