
6.4: Polar Reactions
- Page ID
- 67111
Objectives
After completing this section, you should be able to
- identify the positive and negative ends of the bonds present in the common functional groups.
- explain how bond polarity can be enhanced by the interaction of a functional group with a solvent, metal cation or acid.
- explain how the polarizability of an atom can be an important factor in determining the reactivity of a bond.
- describe the heterolytic bond-breaking process.
- use curved (curly) arrows to indicate the movement of electron pairs during bond breakage and bond formation.
- predict whether a given species (compound or ion) is likely to behave as a nucleophile or as an electrophile.
Make certain that you can define, and use in context, the key terms below.
- electrophile
- nucleophile
- polar reaction
- polarizability
You may wish to review Section 2.1 before you begin this section. The relative electronegativities of the elements shown in the periodic table should already be familiar. Remember that it is the relative electronegativities that are important, not the actual numerical values.
Make sure that you understand the polarity patterns of the common functional groups. Do not try to memorize these polarities; rather, concentrate on why they arise. You will encounter these group polarities so frequently that they will soon become “second nature” to you.
Halogens and the Character of the Carbon-Halogen Bond
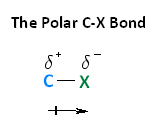
With respect to electronegativity, halogens are more electronegative than carbons. This results in a carbon-halogen bond that is polarized. As shown in the image below, carbon atom has a partial positive charge, while the halogen has a partial negative charge.
The following image shows the relationship between the halogens and electronegativity. Notice, as we move up the periodic table from iodine to fluorine, electronegativity increases.
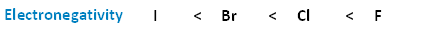
The following image shows the relationships between bond length, bond strength, and molecular size. As we progress down the periodic table from fluorine to iodine, molecular size increases. As a result, we also see an increase in bond length. Conversely, as molecular size increases and we get longer bonds, the strength of those bonds decreases.

The influence of bond polarity
Of the four halogens, fluorine is the most electronegative and iodine the least. That means that the electron pair in the carbon-fluorine bond will be dragged most towards the halogen end. Looking at the methyl halides as simple examples:

The electronegativities of carbon and iodine are equal and so there will be no separation of charge on the bond. One of the important set of reactions of alkyl halides involves replacing the halogen by something else - substitution reactions. These reactions involve either:
- the carbon-halogen bond breaking to give positive and negative ions. The ion with the positively charged carbon atom then reacts with something either fully or slightly negatively charged.
- something either fully or negatively charged attracted to the slightly positive carbon atom and pushing off the halogen atom.
You might have thought that either of these would be more effective in the case of the carbon-fluorine bond with the quite large amounts of positive and negative charge already present. But that's not so - quite the opposite is true! The thing that governs the reactivity is the strength of the bonds which have to be broken. If is difficult to break a carbon-fluorine bond, but easy to break a carbon-iodine one.
The Carbonyl Group
C=O is prone to additions and nucleophillic attack because or carbon's positive charge and oxygen's negative charge. The resonance of the carbon partial positive charge allows the negative charge on the nucleophile to attack the Carbonyl group and become a part of the structure and a positive charge (usually a proton hydrogen) attacks the oxygen. Just a reminder, the nucleophile is a good acid therefore "likes protons" so it will attack the side with a positive charge.
Before we consider in detail the reactivity of aldehydes and ketones, we need to look back and remind ourselves of what the bonding picture looks like in a carbonyl. Carbonyl carbons are sp2 hybridized, with the three sp2 orbitals forming soverlaps with orbitals on the oxygen and on the two carbon or hydrogen atoms. These three bonds adopt trigonal planar geometry. The remaining unhybridized 2p orbital on the central carbonyl carbon is perpendicular to this plane, and forms a ‘side-by-side’ pbond with a 2p orbital on the oxygen.
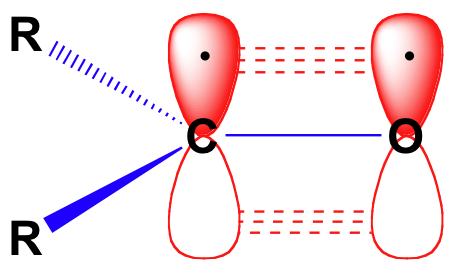
The carbon-oxygen double bond is polar: oxygen is more electronegative than carbon, so electron density is higher on the oxygen side of the bond and lower on the carbon side. Recall that bond polarity can be depicted with a dipole arrow, or by showing the oxygen as holding a partial negative charge and the carbonyl carbon a partial positive charge.

A third way to illustrate the carbon-oxygen dipole is to consider the two main resonance contributors of a carbonyl group: the major form, which is what you typically see drawn in Lewis structures, and a minor but very important contributor in which both electrons in the pbond are localized on the oxygen, giving it a full negative charge. The latter depiction shows the carbon with an empty 2p orbital and a full positive charge.
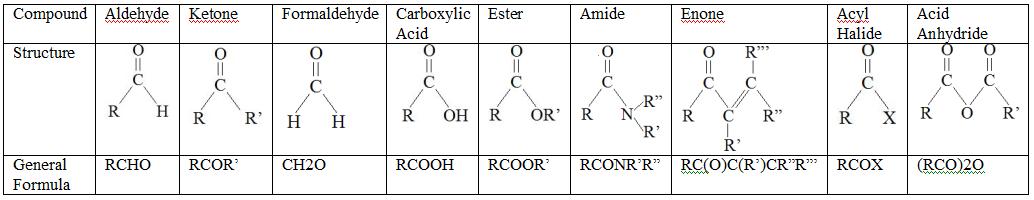
Some Carbonyl Compounds
Nucleophilic Addition to Aldehydes and Ketones
The result of carbonyl bond polarization, however it is depicted, is straightforward to predict. The carbon, because it is electron-poor, is an electrophile: it is a great target for attack by an electron-rich nucleophilic group. Because the oxygen end of the carbonyl double bond bears a partial negative charge, anything that can help to stabilize this charge by accepting some of the electron density will increase the bond’s polarity and make the carbon more electrophilic. Very often a general acid group serves this purpose, donating a proton to the carbonyl oxygen.

The same effect can also be achieved if a Lewis acid, such as a magnesium ion, is located near the carbonyl oxygen. Unlike the situation in a nucleophilic substitution reaction, when a nucleophile attacks an aldehyde or ketone carbon there is no leaving group – the incoming nucleophile simply ‘pushes’ the electrons in the pi bond up to the oxygen.

Alternatively, if you start with the minor resonance contributor, you can picture this as an attack by a nucleophile on a carbocation.

After the carbonyl is attacked by the nucleophile, the negatively charged oxygen has the capacity to act as a nucleophile. However, most commonly the oxygen acts instead as a base, abstracting a proton from a nearby acid group in the solvent or enzyme active site.

This very common type of reaction is called a nucleophilic addition. In many biologically relevant examples of nucleophilic addition to carbonyls, the nucleophile is an alcohol oxygen or an amine nitrogen, or occasionally a thiol sulfur. In one very important reaction type known as an aldol reaction, the nucleophile attacking the carbonyl is a resonance-stabilized carbanion. In this chapter, we will concentrate on reactions where the nucleophile is an oxygen or nitrogen.
- Nucleophilic Addition to Aldehydes and Ketones
- Nucleophilic Substitution of RCOZ (Z = Leaving Group)
- General reaction
- General mechanism
Polarity Patterns in Other Common Functional Groups



Nucleophile?
Nucleophilic functional groups are those which have electron-rich atoms able to donate a pair of electrons to form a new covalent bond. In both laboratory and biological organic chemistry, the most relevant nucleophilic atoms are oxygen, nitrogen, and sulfur, and the most common nucleophilic functional groups are water, alcohols, phenols, amines, thiols, and occasionally carboxylates.
More specifically in laboratory reactions, halide and azide (N3-) anions are commonly seen acting as nucleophiles.
Of course, carbons can also be nucleophiles - otherwise how could new carbon-carbon bonds be formed in the synthesis of large organic molecules like DNA or fatty acids? Enolate ions (section 7.5) are the most common carbon nucleophiles in biochemical reactions, while the cyanide ion (CN-) is just one example of a carbon nucleophile commonly used in the laboratory. Reactions with carbon nucleophiles will be dealt with in chapters 13 and 14, however - in this chapter and the next, we will concentrate on non-carbon nucleophiles.
When thinking about nucleophiles, the first thing to recognize is that, for the most part, the same quality of 'electron-richness' that makes a something nucleophilic also makes it basic: nucleophiles can be bases, and bases can be nucleophiles. It should not be surprising, then, that most of the trends in basicity that we have already discussed also apply to nucleophilicity.

Electrophiles
In the vast majority of the nucleophilic substitution reactions you will see in this and other organic chemistry texts, the electrophilic atom is a carbon which is bonded to an electronegative atom, usually oxygen, nitrogen, sulfur, or a halogen. The concept of electrophilicity is relatively simple: an electron-poor atom is an attractive target for something that is electron-rich, i.e. a nucleophile. However, we must also consider the effect of steric hindrance on electrophilicity. In addition, we must discuss how the nature of the electrophilic carbon, and more specifically the stability of a potential carbocationic intermediate, influences the SN1 vs. SN2 character of a nucleophilic substitution reaction.![]()
Consider two hypothetical SN2 reactions: one in which the electrophile is a methyl carbon and another in which it is tertiary carbon.
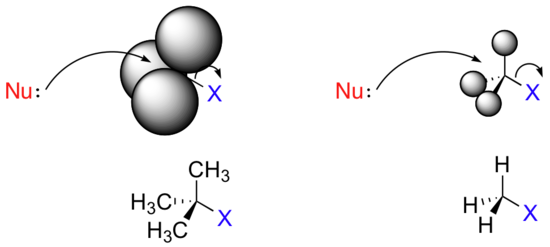
Because the three substituents on the methyl carbon electrophile are tiny hydrogens, the nucleophile has a relatively clear path for backside attack. However, backside attack on the tertiary carbon is blocked by the bulkier methyl groups. Once again, steric hindrance - this time caused by bulky groups attached to the electrophile rather than to the nucleophile - hinders the progress of an associative nucleophilic (SN2) displacement.
The factors discussed in the above paragraph, however, do not prevent a sterically-hindered carbon from being a good electrophile - they only make it less likely to be attacked in a concerted SN2 reaction. Nucleophilic substitution reactions in which the electrophilic carbon is sterically hindered are more likely to occur by a two-step, dissociative (SN1) mechanism. This makes perfect sense from a geometric point of view: the limitations imposed by sterics are significant mainly in an SN2 displacement, when the electrophile being attacked is a sp3-hybridized tetrahedral carbon with its relatively ‘tight’ angles of 109.4o. Remember that in an SN1 mechanism, the nucleophile attacks an sp2-hybridized carbocation intermediate, which has trigonal planar geometry with ‘open’ 120 angles.

With this open geometry, the empty p orbital of the electrophilic carbocation is no longer significantly shielded from the approaching nucleophile by the bulky alkyl groups. A carbocation is a very potent electrophile, and the nucleophilic step occurs very rapidly compared to the first (ionization) step.
Exercise
Label the following either an electrophile or a nucleophile.
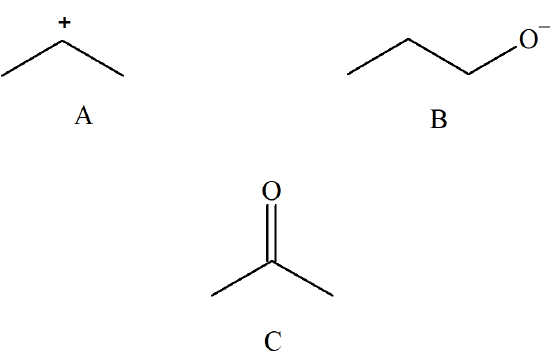
- Answer
-
A = Electrophile
B = Nucleophile
C = Nucleophile
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)