
2. Reversibility – Chemical vs. Electrochemical
- Page ID
- 61288

The term “reversible” is probably the most confusing, misused, and ambiguous term in all of electrochemistry. An attempt will be made here to clearly define the different ways in which the term can be used.
For the reduction of an oxidized form (Ox) of a solution species, the electron transfer can be represented by
\[\ce{Ox + n\: e^{-} <=> Red}\]
where n represents the stoichiometric number of electrons for the process, and Red represents the reduced form of the solution species. The double-sided arrow, common in equilibrium expressions, shows here that the oxidized form of the solution species can be regenerated in a gross chemical sense from the reduced form, meaning that the product is stable on the time scale of the voltammetric experiment. The oxidation reaction can then be expressed by
\[\ce{Red <=> Ox + \mathit{n}\: e^{-}}\]
This example illustrates the condition of chemical reversibility for the Ox/Red couple.
A second kind of reversibility encountered in voltammetry is that of electrochemical reversibility. This refers to the rate at which the electron transfer occurs between the working electrode and the solution redox species. If the transfer occurs quickly without significant thermodynamic barriers, it is called reversible or electrochemically reversible. Electron transfers which are not fast because of one kind of complication or another are referred to as irreversible or electrochemically irreversible. Intermediate rates of electron transfer define the quasi-reversible regime.
By taking a closer look at each of these cases involving reversibility, future confusion can be avoided.
Chemical Reversibility
As we have defined them here, electron transfer is a heterogenous process occurring between a solid working electrode and a solution species close to or at the surface of the electrode. To determine the extent of chemical reversibility, we must ask ourselves if the electrochemically generated species is stable to chemical (homogenous) reaction(s) following the electron transfer. A follow-up chemical reaction can be expressed by
\[\mathrm{Ox + \mathit{n}\: e^- \Leftrightarrow Red \xrightarrow{k_c} Z}\]
with Z being a product which, unlike Red, can no longer be converted back to Ox by direct electron transfer. The magnitude of the chemical rate constant, kc, will determine the extent to which Red reacts to form Z. If kc is large, the redox couple Ox/Red will be chemically irreversible on the time scale of the experiment. If kc is small (or zero), the observed couple will be chemically reversible. For intermediate values of kc, the couple is said to have limited chemical reversibility. Note that the expression quasi-reversible is not used in this context – that is reserved for use in describing electrochemical reversibility.
It should be clear that the observation of chemical reversibility is related to how fast the experiment is conducted. For a measurement that has a slow time scale, a particular system may appear chemically irreversible, while for a faster measurement chemical reversibility for the system may be observed. Voltammetry is well suited to measuring the rate constants leading to product formation, as potential scans at the working electrode can be performed at rates that are slow to those (utilizing ultramicroelectrodes) that are very, very fast.
Electrochemical Reversibility
Electron transfer to solution species at solid electrodes is by nature heterogeneous. As such, reactions of this type have rate constants that depend upon the potential applied to the electrode, with increasing potential leading to an increased rate. Further, the rate at which a solution redox species can undergo electron transfer is dependent upon the concentration of that species at the electrode surface. We will see later how those concentrations change as the potential applied to the electrode changes.
Consider the chemically reversible system we saw before, but now focus on the rate constants for the forward and reverse electron transfer steps, kf and kr.
\[\mathrm{Ox + \mathit{n}\: e^- \overset{k_f}{\underset{k_r}{\Leftrightarrow }} Red}\]
At equilibrium, the forward and reverse rate constants are equal when the applied potential is equal to the E0 of the redox couple. We define the standard heterogeneous rate constant for these conditions, expressed as ks (or k0), and having units of cm/s (resulting from concentration of redox active species in mol/cm3 and electron transfer to an electrode of area expressed in cm2). Units for k Large values for ks indicate that following the application of an applied potential, equilibrium between Ox and Red will be re-established quickly. Small values for ks indicate slow kinetics and longer time requirement for equilibrium. The type and complexity of the molecules undergoing electron transfer, and the molecular rearrangements that may occur following electron transfer are among the many factors that determine the magnitude of ks. Commonly accepted ranges4 for ks are:
| ks > 0.020 cm/s | Reversible |
| 0.020 > ks > 5.0 x 10-5 cm/s | Quasi-reversible |
| ks < 5.0 x 10-5 cm/s | Irreversible |
The forward and reverse rate constants (kf and kb) change as the applied potential, E, moves away from the E0 value, according to
\[\mathrm{k_f = k_s\: exp [-α\: n\: F (E - E^0) / RT]}\]
and
\[\mathrm{k_b = k_s\: exp [(1 - α)\: n\: F\: (E - E^0) / RT]}\]
where α is the transfer coefficient, R is the gas constant (8.314 (V C)/(mol K)) and other variables have their previously defined meaning. α is used to describe the symmetry between the forward and reverse electron transfer steps, according to the Butler-Volmer model of electrode kinetics. If one thinks in terms of energy barriers between the oxidized form of the species and the reduced form as Ox + e- \(\Leftrightarrow \) Red, the free energy of the reactants can be changed by increasing or decreasing the potential applied to the electrode. The extent to which the free energy change contributes to a change in the activation energy is dependent on the magnitude of α, which ranges in value from 0 to 1. This can best be demonstrated graphically (Figure 5), with the steepness of the forward and reverse energy barriers being related to the magnitude of the respective rate constant.
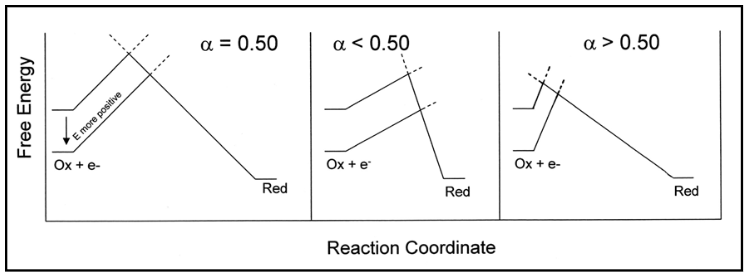
Figure 5
The separation between the traces corresponding to the two values of free energy (and respective values of E) is the same for each value of α shown, while the corresponding change in activation energy for the forward step shows a clear dependence on the magnitude of α. Most electrochemical systems have values of α between 0.3 and 0.7.
Energy over and above the thermodynamic energy (expressed by E0) needed to drive the electron transfer reaction at a certain rate is referred to as overpotential. The result of small ks and related overpotential is to draw out the corresponding voltammetric waves observed for a redox couple. This effect is illustrated in Figure 6 for cyclic voltammetry, a technique in which the current due to a faradaic process is measured as a function of the potential applied to the electrode. Cyclic voltammetry will be further discussed later in the module. Only the value for ks is changed between traces in Figure 6, which were simulated using a program called DigiElch, by Manfred Rudolph.5 Similar effects on peak shape are observed for decreasing values of the transfer coefficient (α) for the forward reaction. The scan rates for each of the three traces shown are the same. Peak shape can also be affected by increasing or decreasing the scan rate.
A fundamental expression for characterizing redox systems that are in the reversible (fast) regime is the Nernst equation. One usually has encountered this expression early in their study of electrochemistry, perhaps in a general chemistry course long ago. The Nernst equation allows the calculation of relative equilibrium concentrations of the two species in a redox pair as a function of the applied electrode potential (E) and the thermodynamic potential (E0) for the pair.
For the general reduction reaction
\[\mathrm{\mathit{a}\, Ox + \mathit{n}\, e^- \Leftrightarrow \mathit{b}\, Red}\]
the Nernst equation takes the form
\[\mathrm{E = E^0 - (RT/nF) \log [A_{Red}^b / A_{Ox}^a]}\]
where R is the gas constant (8.314 (V.C)/(K.mol)), T is temperature in K, n is the stoichiometric number of electrons involved in the process, F is the Faraday constant (96,485 C/equivalent), ARed and AOx are the activities of the reduced and oxidized members of the redox pair, respectively, raised to the appropriate stoichiometric coefficient for each member of the pair. If additional species are involved in the reaction, their activities and stoichiometric coefficients will also appear in the log term of the Nernst equation.
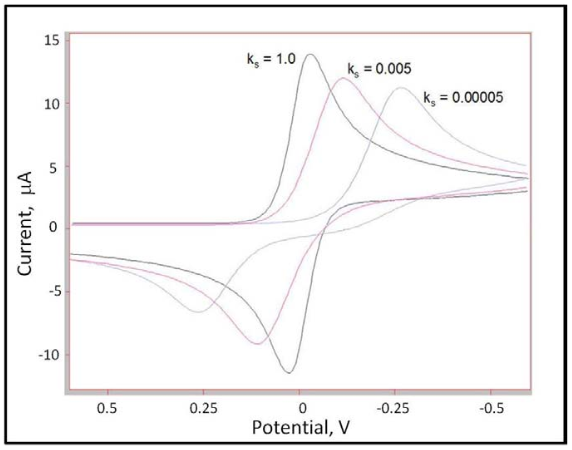
Figure 6
In practical usage, it is convenient to replace activities in the Nernst equation with molar concentrations. Because E0 values are standard reduction potentials of half-cells measured relative to the standard hydrogen electrode (SHE) with all species at unit activity, they are not appropriate when concentration values are used. Instead, it is proper to use formal potentials, E0’, which are half-cell potentials measured relative to SHE when the concentration ratio of Red/Ox is unity and other solution conditions are specified. Substituting E0’ and concentration terms for Red and Ox, the Nernst equation at 25 oC becomes
\[\mathrm{E = E^{0}}\mathrm{' - (0.05916 / n) \log [C_R / C_O]}\]