
3.32: A Numerical Huckel Calculation on Anthracene and Phenanthrene
- Page ID
- 154839

High level quantum mechanical calculations reveal that phenanthrene is more stable than anthracene, and that the enhanced stability has its origin in the energy of the π‐electron density. This tutorial shows that even a simple Huckel molecular orbital calculation predicts the greater stability of phenanthrene.
Huckel calculations can be carried out in several ways; perhaps the most economical and revealing is the numeric method which is based on a connectivity matrix. Anthracene and phenanthrene, shown below, have 14 carbon atoms, so the connectivity matrix is 14 x 14. The only non‐zero matrix elements contain the integer 1 indicating adjacent (bonded) carbons. For example, using the numbering scheme shown below carbon 1 is bonded to carbons 2 and 14 in both molecules, and this connectivity (bonding) is registered in the first row of the respective Huckel matirices shown below.
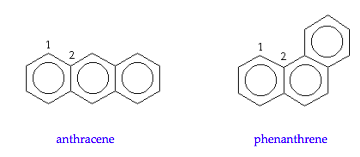
The Huckel connectivity matrices for anthracene and phenanthrene are,
\[ \begin{matrix} Ha = \begin{pmatrix} 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 \\ 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 0 & 0 \\ 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 1 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 1 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 \\ 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 \\ 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 \end{pmatrix} & Hp = \begin{pmatrix} 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 \\ 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 0 & 0 \\ 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 1 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 1 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 & 0 \\ 0 & 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 & 0 \\ 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 & 1 \\ 1 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 0 & 1 & 0 \end{pmatrix} \end{matrix} \nonumber \]
The energy eigenvalues are the negatives of the eigenvalues of the connectivity matrices. The energy eigenvalues, π−electron energy and the π−delocalization energy are calculated for each molecule below. There are seven doubly occupied molecular orbitals for each molecule and the energy of a localized π electron pair (ethene) is ‐2. Thus the π‐electron energy is twice the sum of the occupied energy levels. The delocalization energy is the π‐electron energy minus the energy of an equivalent number of localized π‐electron pairs. See the appendix for the calculation of the ethene (localized) π‐electron energy.
Anthracene
\[ \begin{matrix} \text{Ha = -Ha} & \text{Ea = eigenvals(Ha)} & \text{Ea = sort(Ea)} \end{matrix} \nonumber \]
\[ Ea^T = \begin{pmatrix} -2.414 & -2 & -1.414 & -1.414 & -1 & -1 & -0.414 & 0.414 & 1 & 1 & 1.414 & 1.414 & 2 & 2.414 \end{pmatrix} \nonumber \]
\[ \begin{matrix} E_{ \pi a} = 2 \sum_{i = 1}^7 Ea_i & E_{ \pi a} = -19.314 & Ea_{deloc} = E_{ \pi a} -7(-2) & Ea_{deloc} = -5.314 \end{matrix} \nonumber \]
Phenanthrene
\[ \begin{matrix} \text{Hp = -Hp} & \text{Ep = eigenvals(Hp)} & \text{Ep = sort(Ep)} \end{matrix} \nonumber \]
\[ Ep^T = \begin{pmatrix} -2.435 & -1.951 & -1.516 & -1.306 & -1.142 & -0.769 & -0.605 & 0.605 & 0.769 & 1.142 & 1.306 & 1.516 & 1.951 & 2.414 \end{pmatrix} \nonumber \]
\[ \begin{matrix} E_{ \pi p} = 2 \sum_{i = 1}^7 Ep_i & E_{ \pi p} = -19.448 & Ep_{deloc} = E_{ \pi p} -7(-2) & Ep_{deloc} = -5.448 \end{matrix} \nonumber \]
Summary
\[ \begin{pmatrix} \text{Molecule} & E_{ \pi} & E_{deloc} \\ \text{Anthracene} & -19.314 & -5.314 \\ \text{Phenanthrene} & -19.448 & -5.448 \end{pmatrix} \nonumber \]
According to the Huckel model for π‐electron energy phenanthrene is more stable than anthracene. This result is in agreement with higher level quantum mechanical calculations and the enthalpies of formation of anthracene and phenanthrene, 227 and 201 kJ/mol, respectively.
Appendix
Ethene is the bench mark for a localized pair of π electrons. As shown below, the eigenvalues for the Huckel connectivity matrix for ethene are ‐1 and +1. Thus a localized pair of π electrons has an energy of ‐2.
\[ \begin{matrix} He = \begin{pmatrix} 0 & 1 \\ 1 & 0 \end{pmatrix} & \text{He = -He} & \text{Ee = eigenvals(He)} & \text{Ee = sort(Ee)} & Ee^T = \begin{pmatrix} -1 & 1 \end{pmatrix} \end{matrix} \nonumber \]
References
See problem 14.16 on page 469 of Atkins and de Paula, Physical Chemistry, 7th edition.
For results of high level calculations see, Poater, J. Journal of Organic Chemistry, 2007, 72, 1134.