
3.28: A Numeric Huckel MO Calculation Using Mathcad
- Page ID
- 154423

Enter the number of carbon atoms. \( \text{Natoms} = 4\)
Enter the number of occupied molecular orbitals. \( \text{Nocc} = 2\)
Enter the Huckel matrix.
\[ \begin{matrix} H = \begin{pmatrix} 0 & 1 & 0 & 0 \\ 1 & 0 & 1 & 0 \\ 0 & 1 & 0 & 1 \\ 0 & 0 & 1 & 0 \end{pmatrix} & H = -H \end{matrix} \nonumber \]
Calculate eigenvalues and eigenvectors:
\[ \begin{matrix} E = \text{eigenvals(H)} & \text{Display = rsort} \left( \text{stack} \left( E^T,~ \text{eigenvecs(H)} \right),~ 1 \right) \end{matrix} \nonumber \]
Display eigenvalues and eigenvectors:
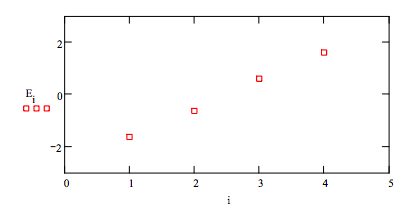
\[ \text{Display} = \begin{pmatrix} -1.618 & -0.618 & 0.618 & 1.618 \\ 0.372 & -0.602 & 0.602 & -0.372 \\ 0.602 & -0.372 & -0.372 & 0.602\\ 0.602 & 0.372 & -0.372 & -0.602 \\ 0.372 & 0.602 & 0.602 & 0.372 \end{pmatrix} \nonumber \]
Display energy level diagram. \( \begin{matrix} \text{E = sort(E)} & \text{i = 1 .. Natoms} \end{matrix}\)
Calculate total π-electronic energy:
\[ \begin{matrix} E_ \pi = 2 \sum_{i = 1} ^{ \text{Nocc}} E_i & E_ \pi = -4.472 \end{matrix} \nonumber \]
Calculate the delocalization energy:
\[ \begin{matrix} E_{deloc} = E_ \pi + 2 \text{Nocc} & E_{deloc} = -0.472 \end{matrix} \nonumber \]
Calculate the delocalization energy per atom:
\[ \frac{ \text{E}_{deloc}}{ \text{Natoms}} = -0.118 \nonumber \]
\[ C = \text{submatrix (Display, 2, Natoms + 1, 1, Natoms)} \nonumber \]
Enter the number of the molecualr orbital to be plotted.
\[ \begin{matrix} r = 1 & s = 1 & 2 \sum_{i=1}^{ \text{Nocc}} \left[ \left( C^{<i>} \right)_r \left( C^{<i>} \right)_s \right] = 1 & \pi- \text{electron density on carbon 1} \\ r = 1 & s = 2 & 2 \sum_{i=1}^{ \text{Nocc}} \left[ \left( C^{<i>} \right)_r \left( C^{<i>} \right)_s \right] = 0.894 & \pi- \text{bond order between carbons 1 and 2} \\ r = 2 & s = 3 & 2 \sum_{i=1}^{ \text{Nocc}} \left[ \left( C^{<i>} \right)_r \left( C^{<i>} \right)_s \right] = 0.447 & \pi- \text{bond order between carbons 2 and 3} \\ r = 3 & s = 4 & 2 \sum_{i=1}^{ \text{Nocc}} \left[ \left( C^{<i>} \right)_r \left( C^{<i>} \right)_s \right] = 1 & \pi- \text{bond order between carbons 3 and 4} \end{matrix} \nonumber \]