
15.4: Standard States for the Fugacity and Activity of a Pure Solid
- Page ID
- 151756

If substance \(A\) is a liquid at one bar and the temperature of interest, pure liquid \(A\) is the standard state for the calculation of the enthalpy and Gibbs free energy of formation. From thermal measurements, we can find the standard Gibbs free energy of formation of this liquid, \({\Delta_fG}^o\left(A,\ell \right)=\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right)\). If we can measure the vapor pressure of the substance and find an equation of state that describes the behavior of the real vapor, we can also find its fugacity and the standard Gibbs free energy of formation of its hypothetical ideal gas, \({\Delta_fG}^o\left(A,{HIG}^o\right)=\mu^o_A\left({HIG}^o\right)\). From the principle that the chemical potential of substance \(A\) is the same in any two phases that are at equilibrium, it follows that the fugacity is the same in each phase.
If we choose the hypothetical ideal-gas standard state as the standard state for the activity of \(A\), then the activity and fugacity are the same thing, and the standard state chemical potential is the same thing as the Gibbs free energy of formation of the hypothetical ideal gas.
\[\mu^o_A={\widetilde\mu}^o_A=\Delta_fG^o\left(A,{HIG}^o\right) \nonumber \]
(activity standard state is the hypothetical ideal gas)
Alternatively, we can choose the pure liquid as the standard state for the activity of \(A\). In this case, there are two further options: We can choose the pure liquid either at one bar pressure, \(P^o\), or at its equilibrium vapor pressure, \(P^{\textrm{⦁}}_{vp}\). If we choose the pure liquid at \(P^o\), we have
\[{\widetilde\mu}^o_A=\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right)=\Delta_fG^o\left(A,{\ell ,P}^o\right) \nonumber \]
(activity standard state is the pure liquid at \(P^o\))
In Chapter 16, we see that the pure liquid at \(P^{\textrm{⦁}}_{vp}\) proves to be the most generally useful choice for \(A\) in a solution. For the pure liquid at \(P^{\textrm{⦁}}_{vp}\), we have
\[{\widetilde\mu}^o_A=\mu^{\textrm{⦁}}_A\left(\ell ,P^{\textrm{⦁}}_{vp}\mathrm{\ }\right) \nonumber \]
(activity standard state is the pure liquid at \(P^{\textrm{⦁}}_{vp}\))
Evidently, it is useful to be able to relate the quantities \(\Delta_fG^o\left(A,{HIG}^o\right)\), \(\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right)=\Delta_fG^o\left(A,{\ell ,P}^o\right)\), and \(\mu^{\textrm{⦁}}_A\left(\ell ,P^{\textrm{⦁}}_{vp}\mathrm{\ }\right)\) to one another.
The difference between the Gibbs free energy of formation of the ideal gas in its hypothetical ideal-gas state and the Gibbs free energy of formation of the liquid in its standard state is a quantity that we can call the standard Gibbs free energy of vaporization, \({\Delta_{vap}G}^o\left(A,P^o\right)\), because both the initial and final states are at a pressure of one bar. At an arbitrary temperature, a liquid at one bar is not at equilibrium with its own ideal gas at one bar, and the standard Gibbs free energy of vaporization is not zero. We have
\[\begin{align*} \Delta_{vap}G^o\left(A,P^o\right) &= \Delta_fG^o\left(A,{HIG}^o\right)-\Delta_fG^o\left(A,{\ell ,P}^o\right) \\[4pt] &= \mu^o_A-\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right) = RT \ln \left[\frac{f_A\left({HIG}^o\right)}{f^{\textrm{⦁}}_A\left(\ell ,P^o\right)}\right] \end{align*} \]
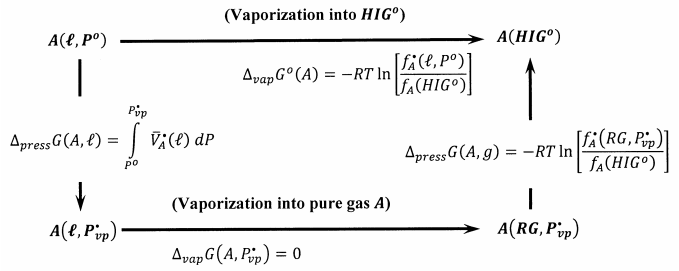
Figure 1 describes a reversible process that takes one mole of \(A\) from the pure liquid state at one bar to its hypothetical ideal-gas standard state. We first reversibly decrease the pressure applied to pure liquid A until we reach its equilibrium vapor pressure, \(P^{\textrm{⦁}}_{vp}\), at the temperature of interest. The molar Gibbs free energy change for this process is
\[\Delta_{press}G\left(A,\ell \right) = \mu^{\textrm{⦁}}_A \left(\ell ,P^{\textrm{⦁}}_{vp}\right)-\Delta_fG^o\left(A,\ell ,P^o\right) = \mu^{\textrm{⦁}}_A\left(\ell ,P^{\textrm{⦁}}_{vp}\right)-\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right) = \int^{P^{\textrm{⦁}}_{vp}}_{P^o}{\overline{V}^{\textrm{⦁}}_A}\left(\ell \right)\ dP \nonumber \]
To reach the hypothetical ideal-gas standard state, we then reversibly and isothermally evaporate one mole of the liquid to its real gas. The Gibbs free energy change for this reversible process is zero, \(\Delta_{vap}G\left(A,P^{\textrm{⦁}}_{vp}\right)=0\). Finally, we isothermally and reversibly expand the real gas to an arbitrarily low pressure, \(P^*\), conceptually convert the real gas to an ideal gas, and compress this ideal gas from \(P^*\) to one bar. The Gibbs free energy change for these latter steps is
\[\begin{align*} \Delta_{press}G\left(A,g\right) &= RT \ln \left[\frac{f_A\left({HIG}^o\right)}{f^{\textrm{⦁}}_A\left(RG,P^{\textrm{⦁}}_{vp}\right)}\right] \\[4pt]&= -RT{ \ln \left[\frac{P^{\textrm{⦁}}_{vp}}{P^o}\right]-\ }RT\int^{P^{\textrm{⦁}}_{vp}}_0{\left[\frac{\overline{V}^{\textrm{⦁}}_A\left(g\right)}{RT}-\frac{1}{P}\right]}dP \end{align*} \]
so that we can also express the standard Gibbs free energy of vaporization as
\[\begin{align*} \Delta_{vap}G^o\left(A,P^o\right) &= \Delta_{press}G\left(A,\ell \right)+\Delta_{press}G\left(A,g\right) \\[4pt]&=\int^{P^{\textrm{⦁}}_{vp}}_{P^o}{\overline{V}^{\textrm{⦁}}_A}\left(\ell \right)\ dP-RT{ \ln \left[\frac{P^{\textrm{⦁}}_{vp}}{P^o}\right]-\ }RT\int^{P^{\textrm{⦁}}_{vp}}_0{\left[\frac{\overline{V}^{\textrm{⦁}}_A\left(g\right)}{RT}-\frac{1}{P}\right]}dP \end{align*} \]
Equating expressions for \({\Delta_{vap}G}^o\left(A,P^o\right)\), we find
\[\begin{align*} \Delta_fG^o\left(A,{HIG}^o\right)-{\Delta_fG}^o\left(A,\ell ,P^o\right) &= RT \ln \left[\frac{f_A\left({HIG}^o\right)}{f^{\textrm{⦁}}_A\left(\ell ,P^o\right)}\right] \\[4pt]&= \int^{P^{\textrm{⦁}}_{vp}}_{P^o}{\overline{V}^{\textrm{⦁}}_A}\left(\ell \right)\ dP-RT{ \ln \left[\frac{P^{\textrm{⦁}}_{vp}}{P^o}\right]-\ }RT\int^{P^{\textrm{⦁}}_{vp}}_0{\left[\frac{\overline{V}^{\textrm{⦁}}_A\left(g\right)}{RT}-\frac{1}{P}\right]}dP \end{align*} \]
Thus, we can find the standard Gibbs free energy of formation of the hypothetical ideal gas from the standard Gibbs free energy of formation of the liquid, the equilibrium vapor pressure of the pure substance, and the equation of state of the pure gas:
\[ \begin{align*} \Delta_fG^o\left(A,{HIG}^o\right)-\Delta_fG^o\left(A,\ell ,P^o\right) &=\Delta_fG^o\left(A,{HIG}^o\right)-\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right) \\[4pt] &=\int^{P^{\textrm{⦁}}_{vp}}_{P^o}{\overline{V}^{\textrm{⦁}}_A}\left(\ell \right)\ dP-RT{ \ln \left[\frac{P^{\textrm{⦁}}_{vp}}{P^o}\right]-\ }RT\int^{P^{\textrm{⦁}}_{vp}}_0{\left[\frac{\overline{V}^{\textrm{⦁}}_A\left(g\right)}{RT}-\frac{1}{P}\right]}dP \end{align*} \]
If the vapor of the substance behaves as an ideal gas, the last integral vanishes. If we also neglect the integral of the molar volume of the liquid, we have
\[{\Delta_fG}^o\left(A,{HIG}^o\right)={\Delta_fG}^o\left(A,\ell ,P^o\right)-RT{ \ln \left[\frac{P^{\textrm{⦁}}_{vp}}{P^o}\right]\ }=\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right)-RT{ \ln \left[\frac{P^{\textrm{⦁}}_{vp}}{P^o}\right]\ } \nonumber \] (ideal gas)
and \(f^{\textrm{⦁}}_A\left(\ell ,P^o\right)=P^{\textrm{⦁}}_{vp}\).
In this development, we suppose that we know \({\Delta_fG}^o\left(A,\ell ,P^o\right)\) from thermal measurements. We can calculate the Gibbs free energy difference between the liquid and hypothetical ideal-gas standard states if we have an equation of state for the vapor. The chemical potential in the hypothetical ideal-gas standard state is
\[\mu^o_A\left({HIG}^o\right)={\Delta_fG}^o\left(A,{HIG}^o\right) \nonumber \]
and the chemical potential of the pure liquid
\[\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right)={\Delta_fG}^o\left(A,\ell ,P^o\right) \nonumber \]
is expressed as a function of the fugacity of the pure liquid:
\[\mu^{\textrm{⦁}}_A\left(\ell ,P^o\right)=\mu^o_A\left({HIG}^o\right)+RT{ \ln \left[\frac{f^{\textrm{⦁}}_A\left(\ell ,P^o\right)}{f_A\left({HIG}^o\right)}\right]\ } \nonumber \]
The activity formalism provides an alternative way to express the same information. When we choose the pure liquid at one bar as the activity standard state; we set \({\tilde{a}}_A\left(\ell ,P^o\right)=1\). For this activity scale, the standard chemical potential becomes the Gibbs free energy of formation of the pure liquid, \({\widetilde\mu}^o_A\left(\ell \right)={\Delta_fG}^o\left(A,\ell ,P^o\right)\). Since the chemical potential of the hypothetical ideal-gas standard state is \(\mu^o_A\left({HIG}^o\right){{=\Delta }_fG}^o\left(A,{HIG}^o\right)\), the activity relationship becomes
\[{\Delta_fG}^o\left(A,{HIG}^o\right)-{\Delta_fG}^o\left(A,\ell ,P^o\right) =\mu^o_A\left({HIG}^o\right)-{\widetilde\mu}^o_A\left(\ell \right)=-\left(\mu^{\textrm{⦁}}_A\left({\ell ,P}^o\right)-\mu^o_A\left({HIG}^o\right)\right) =RT{ \ln \left[{\tilde{a}}_A\left({HIG}^o\right)\right]\ } \nonumber \]
Comparison with the previous equation shows that \[{ \ln \left[{\tilde{a}}_A\left({HIG}^o\right)\right]\ }={ \ln \left[\frac{f_A\left({HIG}^o\right)}{f^{\textrm{⦁}}_A\left(\ell ,P^o\right)}\right]\ } \nonumber \]
If the pure liquid is at equilibrium with a gas mixture at \(P^o\) in which the mole fraction of \(A\) is \(x^{eq}_A\), the fugacity of the pure liquid is equal to the fugacity of the gas in the mixture; that is,
\[f^{\textrm{⦁}}_A\left(\ell ,P^o\right)=f_A\left(RG,x^{eq}_A,P^o\right) \nonumber \]
so that
\[ \ln \left[{\tilde{a}}_A\left({HIG}^o\right)\right] = \ln \left[\frac{f_A\left({HIG}^o\right)}{f_A\left(RG,x^{eq}_A,P^o\right)}\right] \nonumber \]
and
\[{\tilde{a}}_A\left({HIG}^o\right)=\frac{f_A\left({HIG}^o\right)}{f_A\left(RG,x^{eq}_A,P^o\right)} \nonumber \]
Finally, let us take the activity standard state to be pure liquid \(A\) at 1 bar and find the activity of \(A\) in an arbitrary real-gas mixture whose pressure is \(P\) and in which the mole fraction of \(A\) is \(x_A\). Let us represent this state as \(A\left(RG,x_A,P\right)\). The activity of real gas \(A\) in this state is
\[{\tilde{a}}_A\left(RG,x_A,P\right)=\frac{f_A\left(RG,x_A,P\right)}{f^{\textrm{⦁}}_A\left(\ell ,P^o\right)} = \frac{f_A\left(RG,x_A,P\right)}{f_A\left(RG,x^{eq}_A,P^o\right)} \nonumber \]
and the chemical potential is
\[\mu_A\left(RG,x_A,P\right)={\widetilde\mu}^o_A\left(\ell \right)+RT{ \ln \left[{\tilde{a}}_A\left(RG,x_A,P\right)\right]\ } \nonumber \]
If the real gas that is present at mole fraction \(x_A\) in a system whose pressure is \(P\) can be treated approximately as an ideal gas, these gas-fugacity terms can be approximated as
\[f_A\left(RG,x_A,P\right)\approx f_A\left(IG,x_A,P\right)=x_AP \nonumber \] and \[f_A\left(RG,x^{eq}_A,P^o\right)\approx f_A\left(IG,x^{eq}_A,P^o\right)=f_A\left(IG,P^{\textrm{⦁}}_{vp}\right)=P^{\textrm{⦁}}_{vp} \nonumber \]
The activity becomes
\[{\tilde{a}}_A\left(RG,x_A,P\right)\approx {\tilde{a}}_A\left(IG,x_A,P\right)\approx {x_AP}/{P^{\textrm{⦁}}_{vp}} \nonumber \]
and the chemical potential becomes
\[\mu_A\left(RG,x_A,P\right)\approx \mu_A\left(IG,x_A,P\right)={\widetilde\mu}^o_A\left(\ell \right)+RT{ \ln \left(\frac{x_AP}{P^{\textrm{⦁}}_{vp}}\right)\ } \nonumber \]