
11.10: The E1 and E1cB Reactions
- Page ID
- 31515
After completing this section, you should be able to
- write the mechanism for a typical E1 reaction.
- explain why E1 elimination often accompanies SN1 substitution.
- write an equation to describe the kinetics of an E1 reaction.
- discuss the stereochemistry of E1 reactions.
- account for the lack of a deuterium isotope effect in E1 reactions.
Make certain that you can define, and use in context, the key terms below.
- E1 reaction
- E1cB reaction
The abbreviation E1 stands for “unimolecular elimination”; that is, an E1 reaction is an elimination reaction in which only one species is involved in the rate-limiting step.
Unimolecular Elimination (E1) is a reaction in which the removal of an HX substituent results in the formation of a double bond. It is similar to a unimolecular nucleophilic substitution reaction (SN1) in various ways. One being the formation of a carbocation intermediate. Also, the only rate determining (slow) step is the dissociation of the leaving group to form a carbocation, hence the name unimolecular. Thus, since these two reactions behave similarly, they compete against each other. Many times, both these reactions will occur simultaneously to form different products from a single reaction. However, one can be favored over another through thermodynamic control. Although Elimination entails two types of reactions, E1 and E2, we will focus mainly on E1 reactions with some reference to E2.
General E1 Reaction
An E1 reaction involves the removal of the halogen leaving group followed by the deprotonation of an adjacent hydrogen to produce an alkene product. In order to accomplish this, a Lewis base is required.

Mechanism
This mechanism starts with the spontaneous removal of the leaving group. The leaving group also removes the electrons from the C-Br bond, making the attached carbon a carbocation. In much the same fashion as the SN1 mechanism, the first step of the E1 mechanism is slow thus making it the rate determining step. This makes E1 reaction kinetics unimolecular and first-order with respect to the substrate. Next, a Lewis Base (B-) deprotonates an adjacent hydrogen from the carbocation. The electrons of the C-H bond are donated to the adjacent C-C bond, forming a double bond. Unlike E2 reactions, which require the adjacent proton to be anti to the leaving group, E1 reactions only require a neighboring hydrogen. This is due to the fact that the leaving group has left the molecule to form an achiral, trigonal planar carbocation.

Evidence for the E1 Mechanism
The primary evidence in support of the E1 mechanism is the fact that E1 reaction follow first-order kinetics which is consistent with the reaction mechanism containing a unimolecular dissociation as the rate determining step. Another piece of evidence is seen in the fact that E1 reactions show no deuterium isotope effect. Experiments have shown that there is no difference in reaction rate when deuterated and nondeuterated substrates are used. This is consistent with the E1 mechanism because a C-H bond is not broken in the rate determining step.
Lastly, because the halogen and hydrogen are removed in different steps, the mechanism of E1 reactions predicts that they will not require the anti-periplanar geometry required for E2 reactions. E1 reactions do not have the geometric constraints of E2 reactions discussed in Sections 11-8 and 11-9. In Section 11-9 an example showed that trans-1-Chloro-2-methylcyclohexane was capable of only forming the product 3-methylhexene during an E2 reaction due to the anti-periplanar constraints. When the same substrate is reacted under E1 conditions two elimination products (1-methylhexene and 3-methylhexene) are produced. In the substrate, the hydrogen marked as green cannot obtain anti-periplanar geometry with the Cl leaving group. Under E2 conditions this hydrogen and the Cl cannot be eliminated to form an elimination product. However, under E1 conditions the hydrogen and the Cl eliminate, despite the lack of anti-periplanar geometry, to form 1-methylhexene.

Alkyl Halide Reactivity in E1 Reactions
Due to the fact that E1 reactions create a carbocation intermediate, reactivity of alkyl halides toward E1 reaction mirror those present in SN1 reactions.
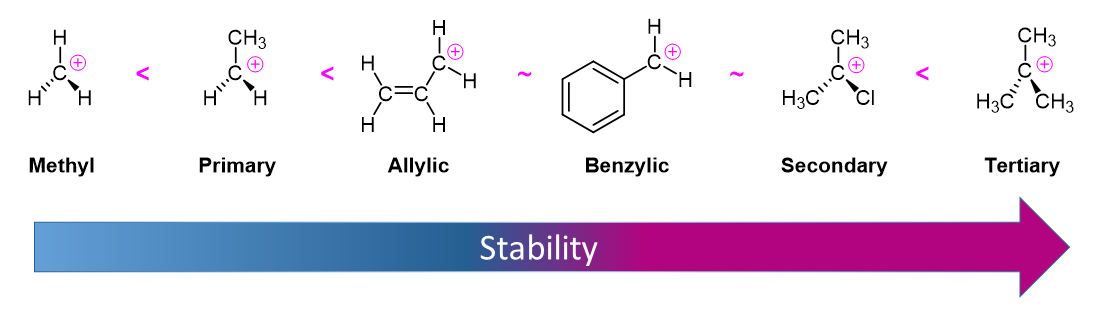
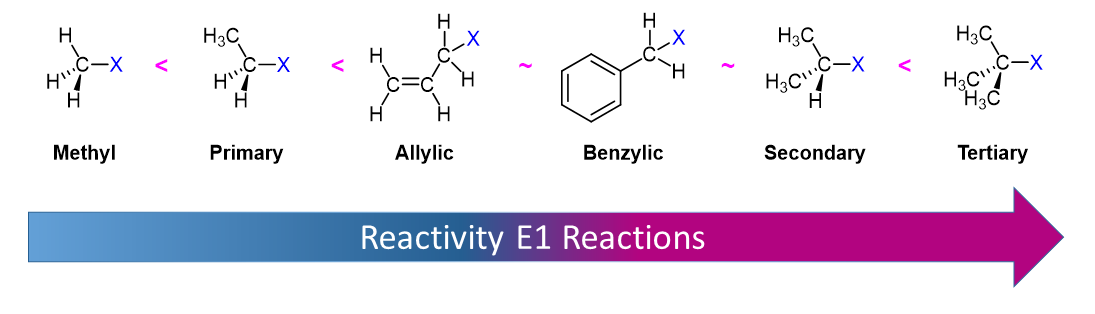
As expected, tertiary carbocations are favored over secondary, primary and methyl carbocations. This is due to the phenomena of hyperconjugation, which essentially allows a nearby C-C or C-H bond to interact with the p orbital of the carbon to bring the electrons down to a lower energy state. Thus, this has a stabilizing effect on the molecule as a whole. In general, primary and methyl carbocations do not proceed through the E1 pathway for this reason, unless there is a means of carbocation rearrangement to move the positive charge to a nearby carbon that is more stable. Secondary and Tertiary carbons form more stable carbocations, thus E1 reactions occur quite rapidly at these atoms.
Secondary carbocations can be subject to the E2 reaction pathway, but this generally occurs in the presence of a good/strong base. Adding a weak base to the reaction disfavors E2, essentially pushing towards the E1 pathway. In many instances, solvolysis occurs rather than using a base to deprotonate. This means heat is added to the solution, and the solvent itself deprotonates a hydrogen. The medium can effect the pathway of the reaction as well. Polar protic solvents may be used to hinder nucleophiles, thus disfavoring E2/SN2 from occurring.
The Connection Between SN1 and E1 Reactions
The E1 and SN1 mechanisms both begin with the rate-determing step, the unimolecular removal of a leaving group to form a carbocation intermediate. Sharing the rate-determining step causes alkyl halides to produce both SN1 substitution and E1 elimination products whenever they are reacted with nonbasic nucleophiles in a protic solvent. For example, the hydrolysis of tert-butyl chloride in a solution of water and ethanol gives a mixture of 2-methyl-2-propanol (60%) and 2-methylpropene (40%) at a rate independent of the water concentration.

To produce the alcohol product, water attacks the carbocation as a nucleophile as part of an SN1 reaction. To produce the alkene product, water acts as a base and deprotonates an adjacent hydrogen as part of an E1 reaction. As expected, the mechanism of the two reactions have similar characteristics. They both show first order kinetics; neither is much influenced by a change in the nucleophile/base; and both are relatively non-stereospecific.

To summarize, when carbocation intermediates are formed one can expect them to react further by one or more of the following modes:
- The cation may bond to a nucleophile to give a substitution product.
- The cation may transfer a adjacent proton to a base, giving an alkene product.
- The cation may rearrange to a more stable carbocation, and then react by mode #1 or #2.
Comparing E1 and E2 Mechanisms
When considering whether an elimination reaction is likely to occur via an E1 or E2 mechanism, we really need to consider three factors:
1) The base: strong bases favor the E2 mechanism, whereas, E1 mechanisms only require a weak base.
2) The solvent: good ionizing solvents (polar protic) favor the E1 mechanism by stabilizing the carbocation intermediate.
3) The alkyl halide: primary alkyl halides have the only structure useful in distinguishing between the E2 and E1 pathways. Since primary carbocations do not form, only the E2 mechanism is possible.
Ultimately, whether the elimination mechanism is E1 or E2 is not very important, since the product is the same alkene. We need to remember, however, that Zeitzev´s rule always determines the most likely alkene to be formed.
| Reaction Parameter | E2 | E1 |
| alkyl halide structure | tertiary > secondary > primary | tertiary > secondary >>>> primary |
| nucleophile | high concentration of a strong base | weak base |
| mechanism | 1-step | 2-step |
| rate limiting step | bimolecular transition state | carbocation formation |
| rate law | rate = k[R-X][Base] | rate = k[R-X] |
| solvent | not important | polar protic |
The E1cB Reaction
Although E1 reactions typically involve a carbocation intermediate, the E1cB reaction utilizes a carbanion intermediate. A proton adjacent to a carbonyl group is removed using a strong base. This proton is acidic because the resulting conjugate base anion is stabilized by delocalization on to the carbonyl group. This anion causes the expulsion of an adjacent leaving group to create an alkene which is conjugated with the carbonyl which is called an enone. This reaction is generally utilized when a poor leaving group, such as a hydroxide, is involved. This poor leaving group makes the direct E1 or E2 reactions difficult. This reaction is used later in a reaction called an aldol condensation which will be discussed in Section 23-3.
General Reaction
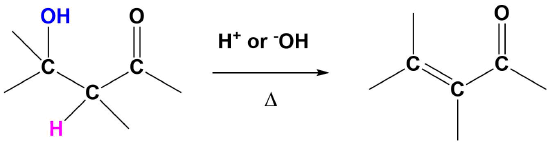
Predicting the Product of an E1cB Reaction


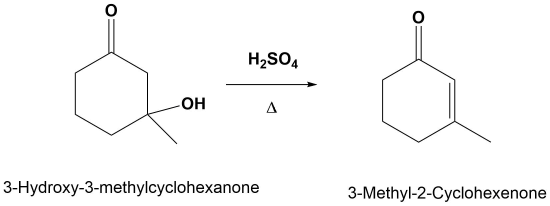
E1cB Mechanism
The mechanism starts with the base removing a hydrogen to form an alkoxide anion. The alkoxide reforms the carbonyl C=O bond promoting the elimination of alcohol OH as a leaving group which also reforms the base catalyst. Although the base catalyzed elimination of alcohols is rare, it happens in this case in part due to the stability of the conjugated enone product.
1) Deprotonation to form the anion

2) Leaving Group Removal

Note! The double bond always forms in conjugation with the carbonyl.
1. Predict the dominant elimination mechanism (E1 or E2) for each reaction below. Explain your reasoning.
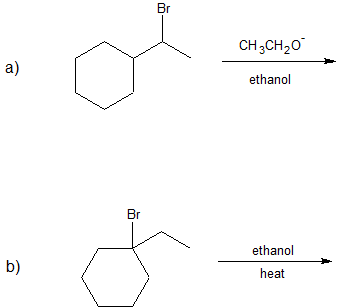
2) Specify the reaction conditions to favor the indicated elimination mechanism.
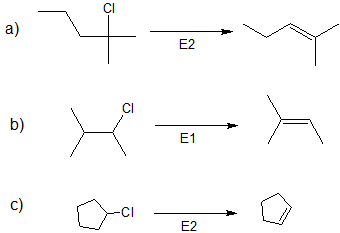
- Answer
-
1)

2)
a) strong base, such as hydroxide, an alkoxide, or equivalent
b) water or alcohol or equivalent weak base with heat
c) strong base, such as hydroxide, an alkoxide, or equivalent