
2.6: Anionic Polymerization
- Page ID
- 238855

If you have studied some organic chemistry, then it seems obvious that cationic methods would be employed for the polymerisation of alkenes. After all, alkenes are nucleophilic, and so they should react readily with cationic initiators. It might be surprising that there are a number of alkene polymerisations that work very well using anionic initiators.
One of the most common scenarios for this approach is in the polymerisation of conjugated hydrocarbons, such as styrene. From the point of view of Lewis structures, this seems like a funny combination. There is no obvious electrophile in an alkene; not even in a conjugated one. Why would it interact with an anionic initiator?

A molecular orbital treatment of conjugated systems provides an answer. When alkenes are conjugated, several things happen. There is at least one extended π-bonding orbital that moves to lower energy because of conjugation, and we think of that factor to explain the relative stability of conjugated alkenes toward electrophiles, compared to the reactivity of isolated alkenes. At the same time, there are also π-bonding orbitals that are raised a little in energy as well as corresponding π*- or π-antibonding orbitals that are lowered in energy by conjugation. This feature explains the well-known shift in optical absorption of conjugated alkenes toward the visible region of the spectrum; we see that in brightly coloured compounds such as carotene and xanthophylls in autumn leaves. It is partly that lowering of the Lowest Unnocupied Molecular Orbital (LUMO) that leads to the increased electophilicity of conjugated alkenes compared to regular alkenes.
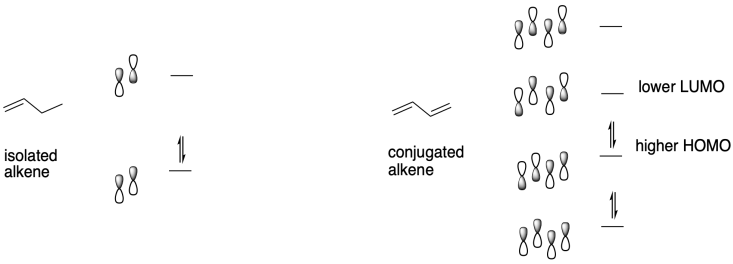
Once an anionic initiator has added to a conjugated alkene, the conjugated alkene itself becomes an anion. That leads to the second part of the explanation. If you look at the anion that results, you will find resonance stabilisation. That delocalisation is always a powerful factor that allows ions to form more easily. In fact, we can easily choose an anionic initiator that is more reactive than that delocalised one, so that there is a driving force toward polymer initiation. Alkyllithiums, such as butyllithium, are good candidates.
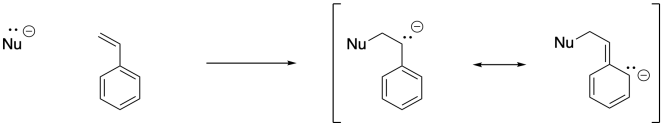
There may be other alkenes that come to mind as being potentially suitable for anionic polymerisation. Again, if you have studied organic chemistry, you might thing of 1,4-addition of nucleophiles to conjugated enones. Anionnic initiation of these kinds of monomers is also possible. Acrylate polymerisation, involving monomers that contain a C=C-CO2 unit, is commercially very important.
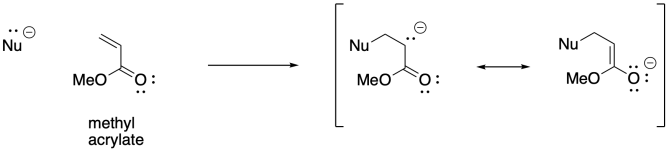
The 1,4-addition of anionic initiators does not proceed as smoothly as we might expect, however. This approach is prone to a number of side reactions. For example, Claisen condensations result in the loss of an alkoxide leaving group and chain death.
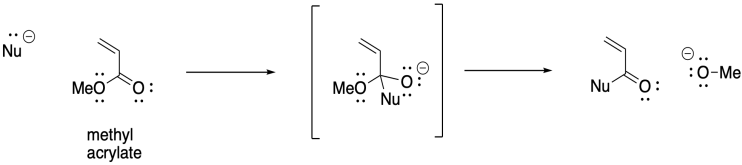
On the other hand, the presence of α-hydrogens along the polymer can lead to a different problem, with equilibrium proton transfer resulting from "back-biting". This phenomenon leads to uncontrolled branching of the polymer chain.
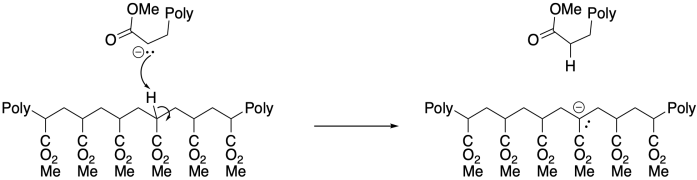
The potential for side reactions means that anionic polymerisations are prone to poor molecular weight control. Chains that die early will have lower molecular weight than those that continue to grow, leading to broad molecular weight distributions. Like cationic polymerisations, anionic polymerisations can be greatly enhanced through the use of living methods.
Problem SM4.1.
Anionic polymerization of p-substituted styrene proceeds very well when the substituent is an electron-withdrawing group such as nitrile. Explain the reason for the success of this approach.
![]()
Problem SM4.2.
Vinyl chloride (CH2=CHCl) might be expected to be a good candidate for anionic polymerization, but side reactions make it unsuitable.
a) Explain why the resulting anion might be expected to show some stability.
b) Show the side reaction that would result under anionic polymerization conditions.