
16.12: Redox reactions involving thiols and disulfides
- Page ID
- 1007

16.12A: Disulfide bridges in proteins
Disulfide (sulfur-sulfur) linkages between two cysteine residues are an integral component of the three-dimensional structure of many proteins. The interconversion between thiols and disulfide groups is a redox reaction: the thiol is the reduced state, and the disulfide is the oxidized state.
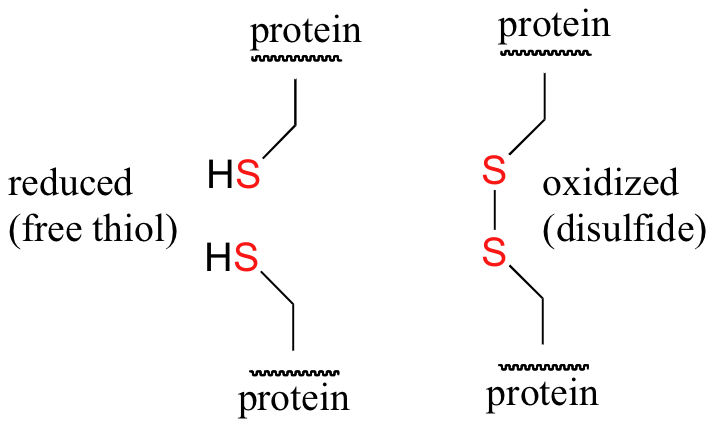
Notice that in the oxidized (disulfide) state, each sulfur atom has lost a bond to hydrogen and gained a bond to a sulfur - this is why the disulfide state is considered to be oxidized relative to the thiol state.
The redox agent that mediates the formation and degradation of disulfide bridges in most proteins is glutathione, a versatile coenzyme that we have met before in a different context (section 14.2A). Recall that the important functional group in glutathione is the thiol, highlighted in blue in the figure below. In its reduced (free thiol) form, glutathione is abbreviated 'GSH'.
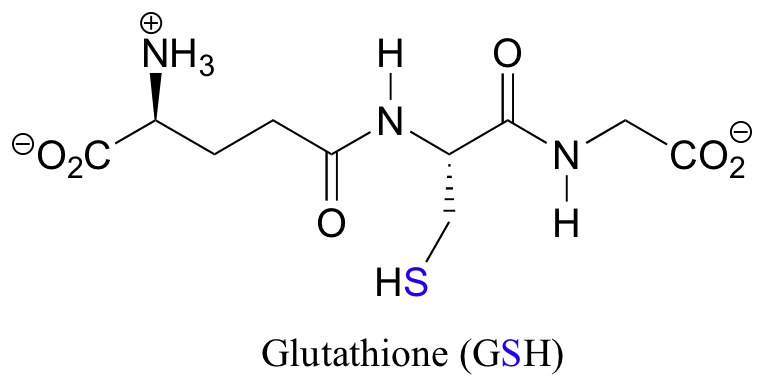
In its oxidized form, glutathione exists as a dimer of two molecules linked by a disulfide group, and is abbreviated 'GSSG'.
A new disulfide in a protein forms via a 'disulfide exchange' reaction with GSSH, a process that can be described as a combination of two SN2-like attacks. The end result is that a new cysteine-cysteine disulfide forms at the expense of the disulfide in GSSG.
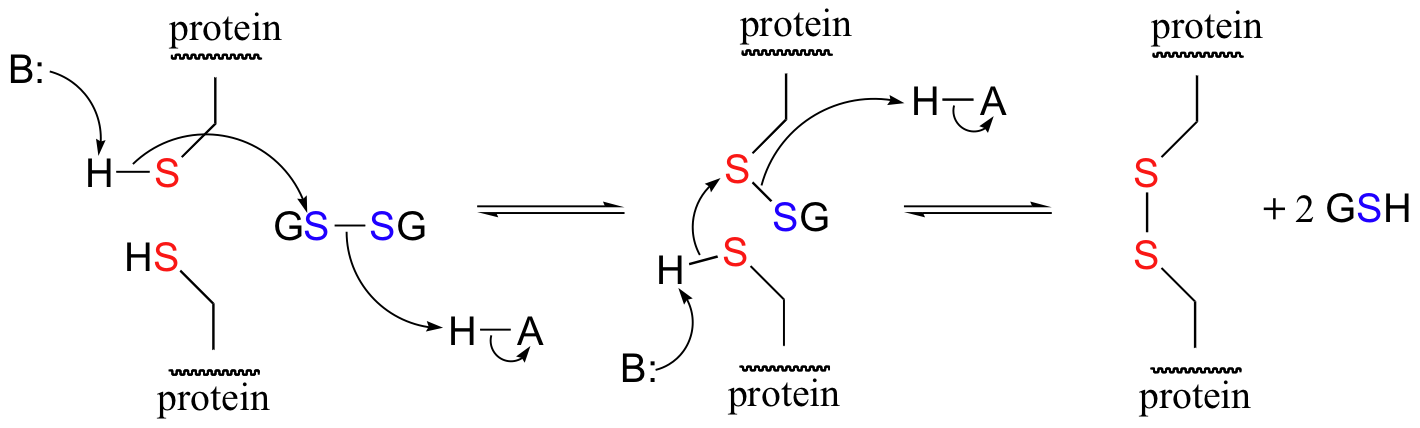
In its reduced (thiol) state, glutathione can reduce disulfides bridges in proteins through the reverse of the above reaction.
Disulfide bridges exist for the most part only in proteins that are located outside the cell. Inside the cell, cysteines are kept in their reduced (free thiol) state by a high intracellular concentration of GSH, which in turn is kept in a reduced state (ie. GSH rather than GSSG) by a flavin-dependent enzyme called glutathione reductase.
Disulfide bridges in proteins can also be directly reduced by another flavin-dependent enzyme called 'thioredoxin'. In both cases, NADPH is the ultimate electron donor, reducing FAD back to FADH2 in each catalytic cycle.
In the biochemistry lab, proteins are often maintained in their reduced (free thiol) state by incubation in buffer containing an excess concentration of b-mercaptoethanol (BME) or dithiothreitol (DTT). These reducing agents function in a manner similar to that of GSH, except that DTT, because it has two thiol groups, forms an intramolecular disulfide in its oxidized form.
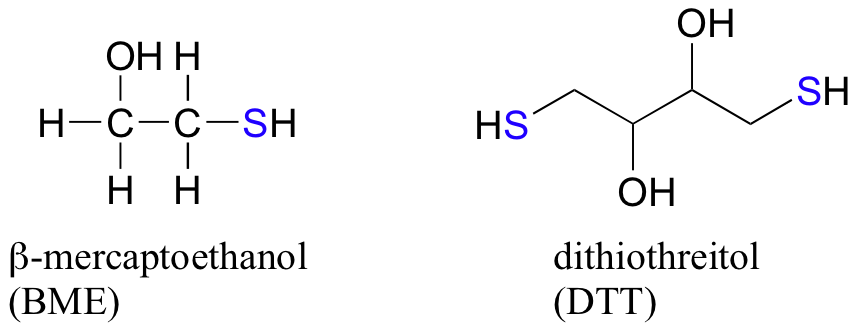
Example 16.12.1
- Draw the structures of the oxidized forms of BME and DTT.
- Draw a mechanism for the reduction of a protein disulfide bond by DTT.
16.12B: The role of disulfides in the pyruvate dehydrogenase reaction
Disulfides play a key role in one of the most important oxidative reactions in central metabolism - the transformation of pyruvate to acetyl CoA by pyruvate dehydrogenase.
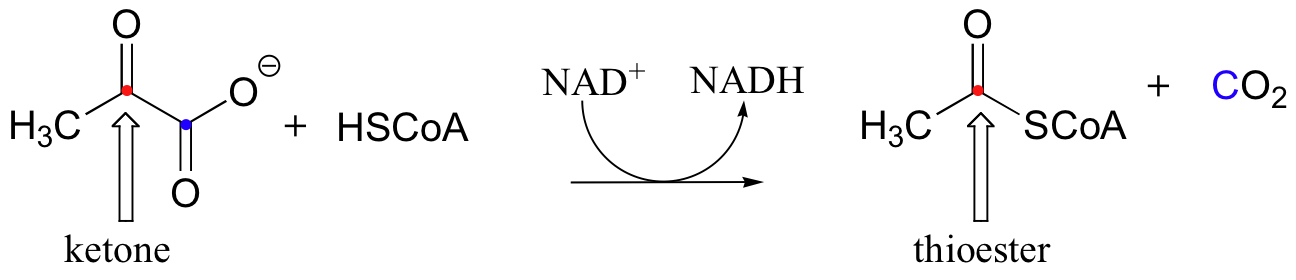
Pyruvate dehydrogenase, because of the many jobs it must carry out, is actually a large, multi-component protein with several active sites. You will learn in your biochemistry course more about the structure of this remarkable enzyme complex, and how this reaction serves as a one-way bridge between glycolysis and the citric acid cycle in animals.
Looking at the reaction, you should be able to recognize that, first of all, the pyruvate substrate is being oxidized - it starts out as a ketone, and ends up as a thioester. Ultimately, the oxidizing agent in this reaction is NAD+, but how the reduction of NAD+ is linked to the oxidative decarboxylation of pyruvate will not become clear until near the end of this section.
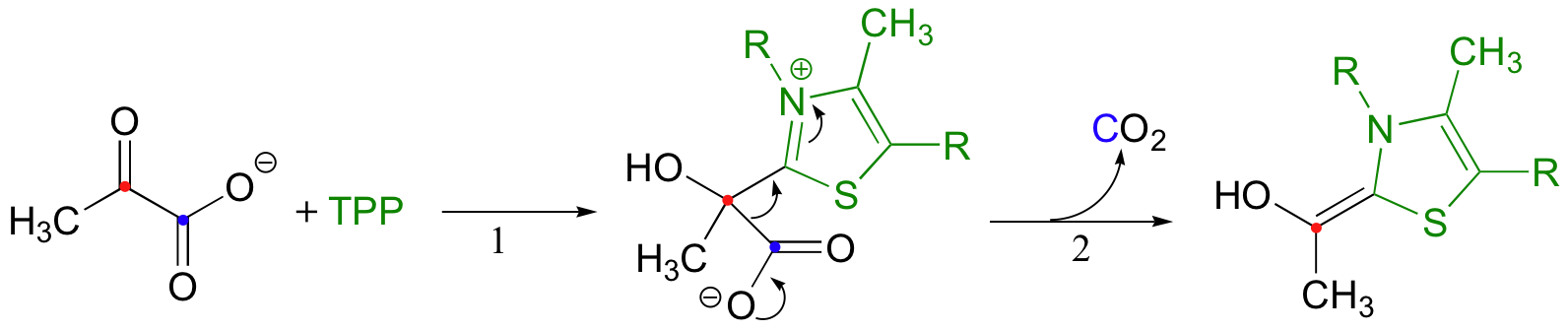
The second thing to notice is that, because a decarboxylation is occurring directly adjacent to a carbonyl, the thiamine diphosphate (TPP) coenzyme will be required.
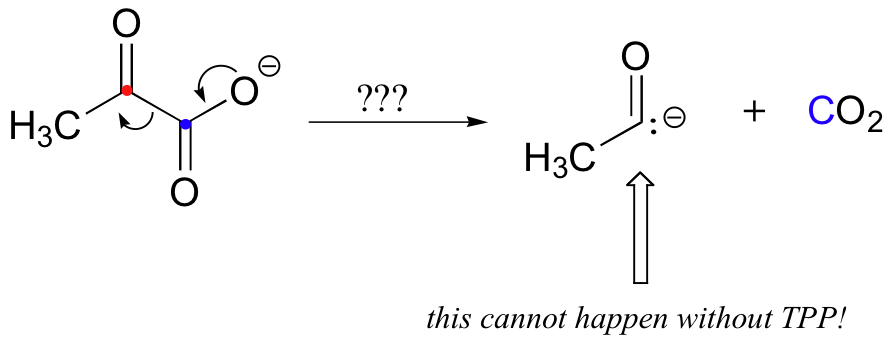
In fact, the first stage of the reaction (steps 1 and 2 below) is identical to that of pyruvate decarboxylase, an enzyme we discussed back in section 14.5C. The TPP coenzyme serves to absorb the electrons from the decarboxylation.
While the TPP-stabilized carbanion intermediate in the pyruvate decarboxylase reaction went on to become protonated, however, the carbanion in the pyruvate dehydrogenase reaction acts as a nucleophile, attacking one of the sulfurs in a disulfide bridge of a coenzyme called lipoamide. (step 3 below). The lipoamide is attached by an amide linkage to one of the enzyme's lysine residues.
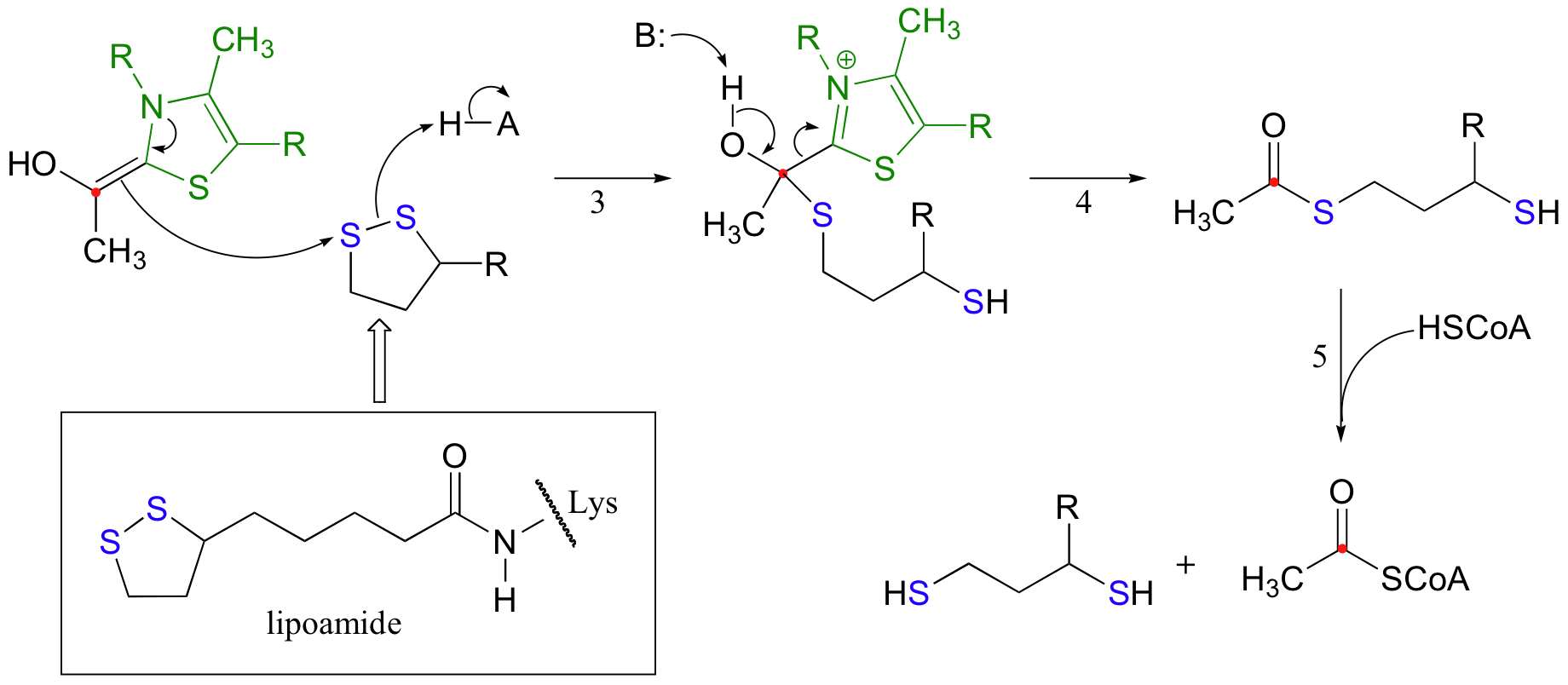
After the usual expulsion of TPP (step 4 above), the result is an acetyl-dihydrolipoamide thioester. This undergoes transesterification (step 5 above) to form acetyl-CoA. So that the enzyme can turn around and catalyze another reaction, the reduced dihydrolipamide is regenerated back to its oxidized state through disulfide exchange with a disulfide bond on the enzyme.
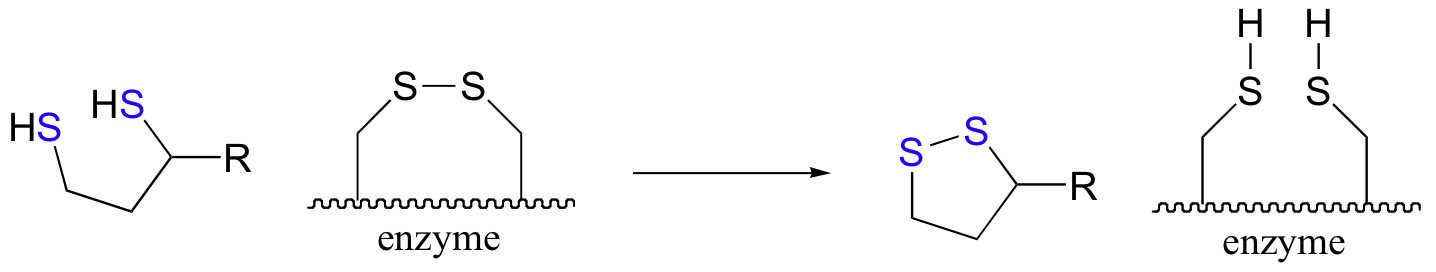
The pair of enzymatic cysteines is then oxidized back to disulfide form by an FAD-dependent reaction that is essentially the reverse of the glutathione reductase reaction described earlier in this section, and finally FAD is regenerated with concurrent reduction of NAD+ .
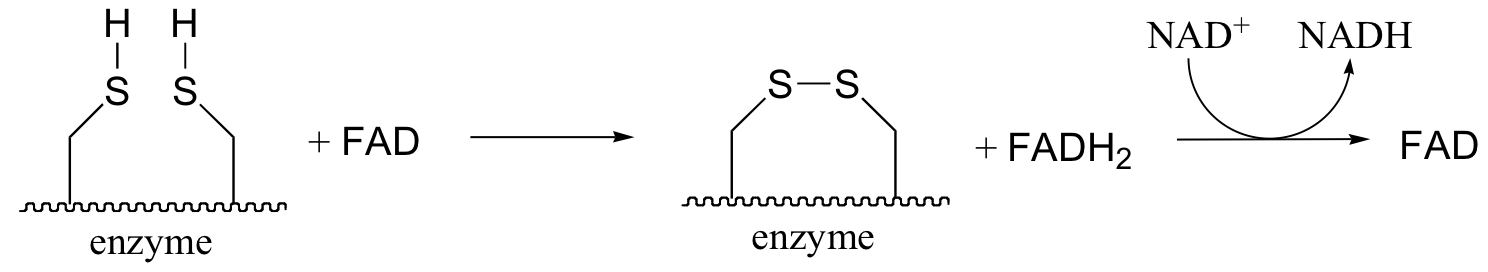
All of this is catalyzed by one enzyme - no wonder it is so big and complex! Overall, the long chain of reactions results in one acetyl CoA being decarboxylated and oxidized (from a ketone to a thioester), and one NAD+ being reduced to NADH.