15.7: Carbocation rearrangements
- Page ID
- 1000

15.7A: Hydride and alkyl shifts
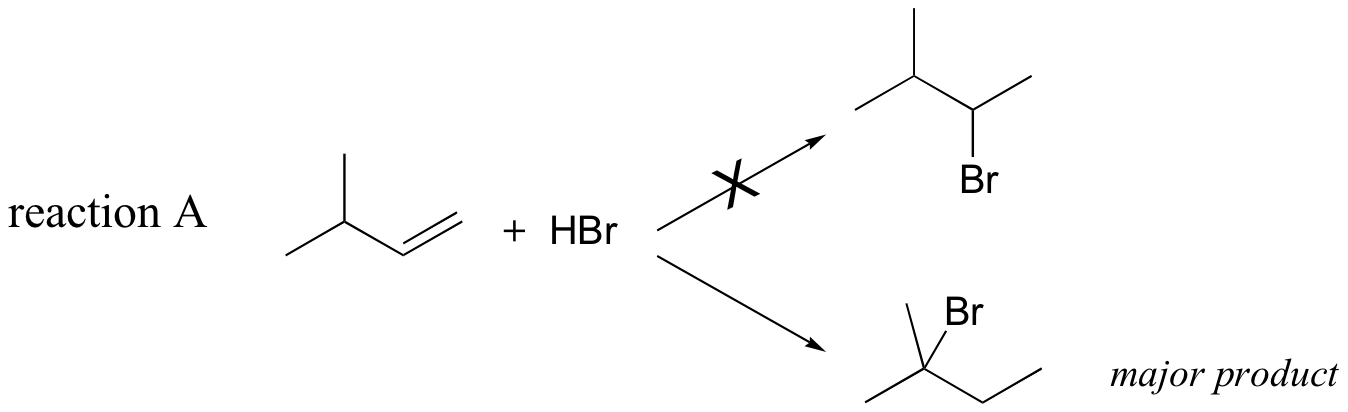
Previously, (section 15.2B) we learned about the so-called 'Markovnikov rule', which can be used to predict the favored regiochemical outcome of electrophilic additions to asymmetric alkenes. According to what we have learned, the addition of HBr to 3-methyl-1-butene should result in a secondary bromoalkane. However, the predominant product that is would actually be observed in this reaction is a tertiary bromoalkane!
Likewise, if HBr is added to 3,3-dimethyl-1-butene, the product is a tertiary - rather than a secondary - bromoalkane:
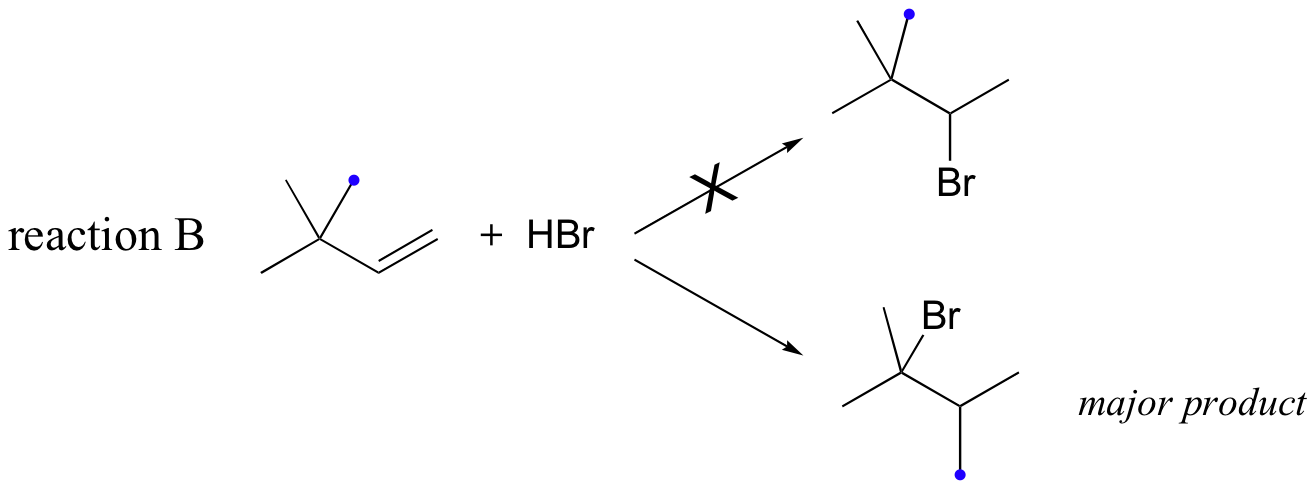
Notice in reaction B that, in the observed product, the carbon framework has been rearranged. Clearly, something is happening in these transformations that cannot be explained by what we have learned so far about electrophilic additions and the nature of carbocation intermediates.
Let's look again at reaction A above in order to see what is happening. Upon protonation of the double bond, a secondary carbocation is formed (step 1).
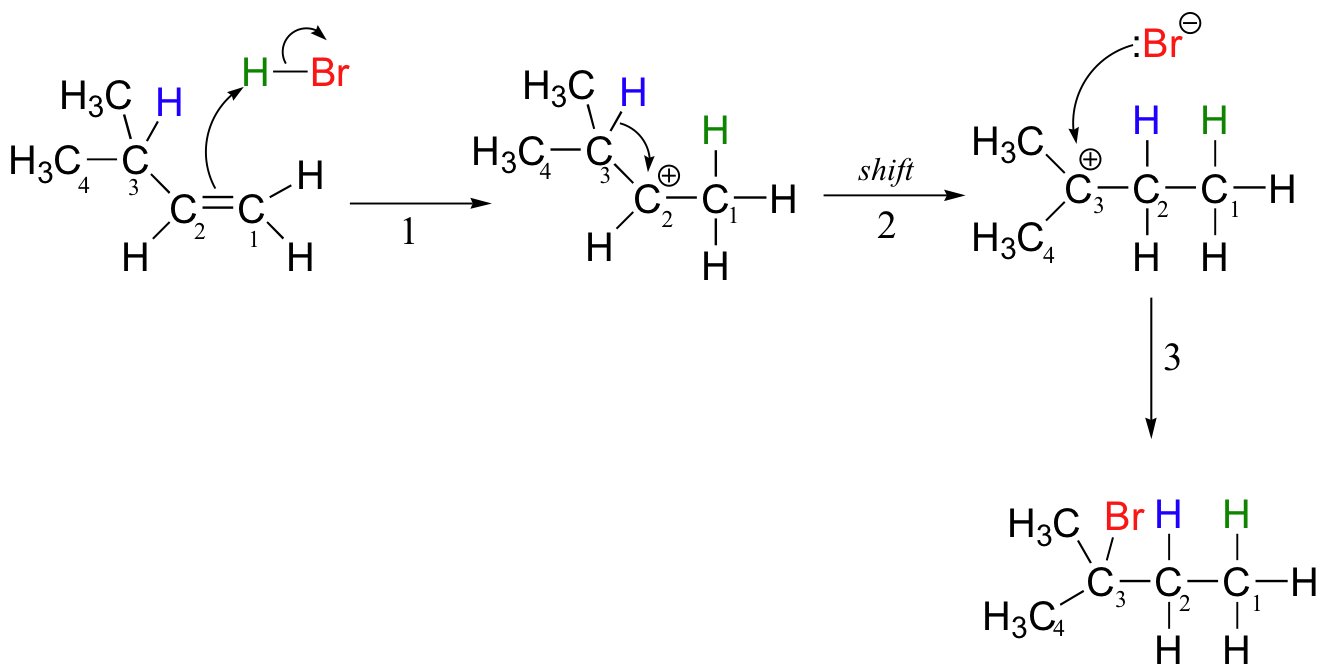
What happens next (step 2 above) is a process called a carbocation rearrangement. The electrons in the bond between C3 and its hydrogen are attracted by the positive charge next door on C2, and so they simply 'shift' over to the empty p orbital on C2, pulling the proton over with them. This step is specifically referred to as a 1-2 hydride shift: the hydride ‘shifts’ one carbon over on the chain. As the shift proceeds, a new C2-H s bond is formed, and C3 is left with an empty orbital and a positive charge. No discrete :H- ion is formed in this process, rather it is a concerted movement of a hydride - two electrons and a proton - from one carbon to another.
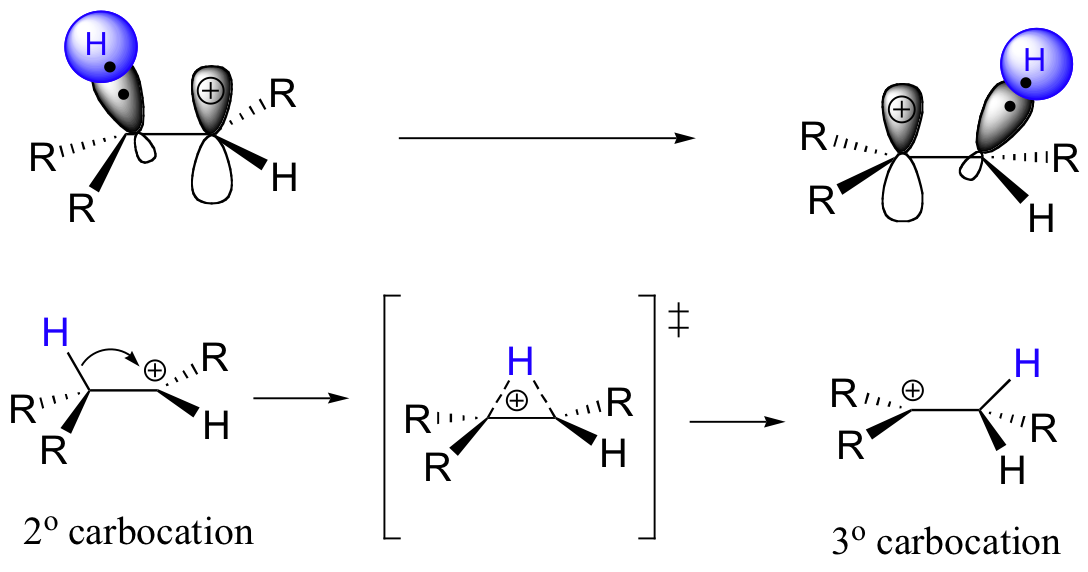
What is the thermodynamic driving force for this shifting process? Notice that the hydride shift results in the conversion of a secondary (2o) carbocation to a (more stable) tertiary (3o) carbocation. It is thus a thermodynamically downhill reaction, and, as it turns out, the energy barrier to the shift is relatively low, meaning that it can occur quite rapidly.
After the hydride shift is complete, the bromide nucleophile is free to attack the new tertiary carbocation to form the observed tertiary bromoalkane product (step 3 in the HBr addition mechanism shown above).
Let's now move to the second hydrobromide addition reaction (reaction B).
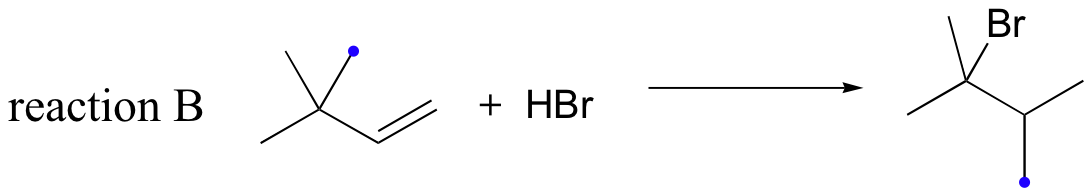
In this case, once the secondary carbocation intermediate is formed, there is no hydrogen on C3 to shift over.
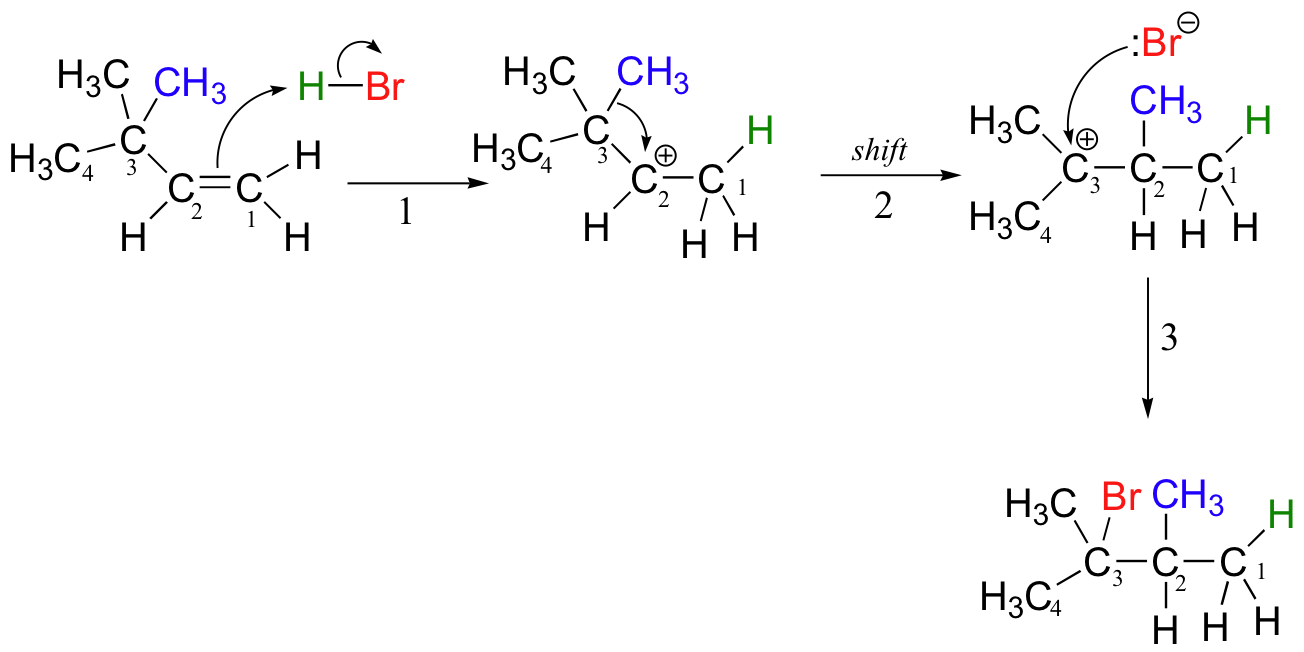
Instead, it is a methyl group rather than a hydride that does the shifting, as the electrons this C-C s-bond move over to fill the empty orbital on C2 (step 2 above). Again, this results in the conversion of a secondary carbocation to a more stable tertiary carbocation, and although a 1-2 methyl shift is slightly slower than a hydride shift, it is still quite rapid. The end result is a rearrangement of the carbon framework of the molecule.
In most examples of carbocation rearrangements that you are likely to encounter, the shifting species is a hydride or methyl group. However, pretty much any alkyl group is capable of shifting. Sometimes, the entire side of a ring will shift over in a ring-expanding rearrangement. Consider the following electrophilic addition of HBr to an alkene: right off the bat, it is very hard to see what is going on here.
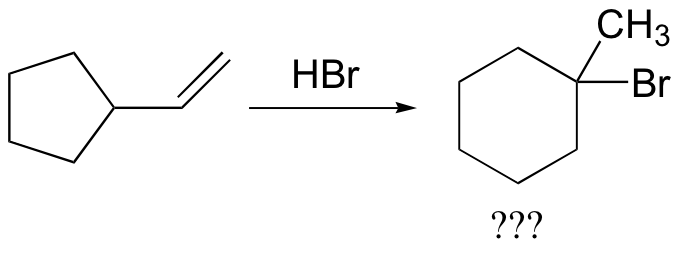
Taking into account the possibility of rearrangement, however, we can see how the observed product forms.

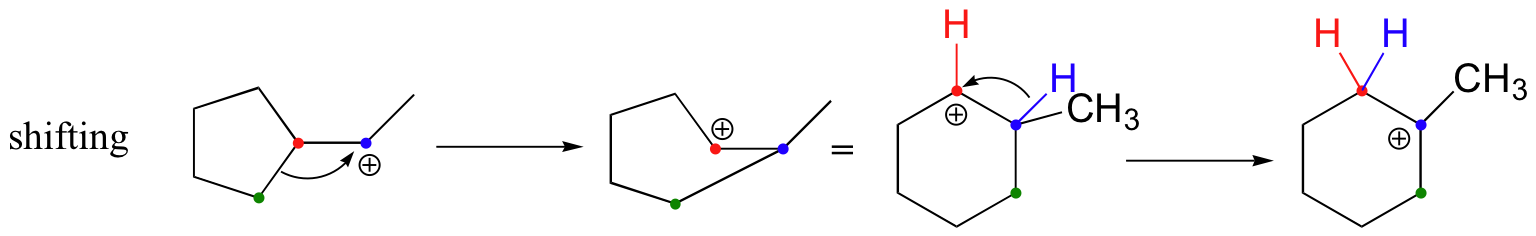
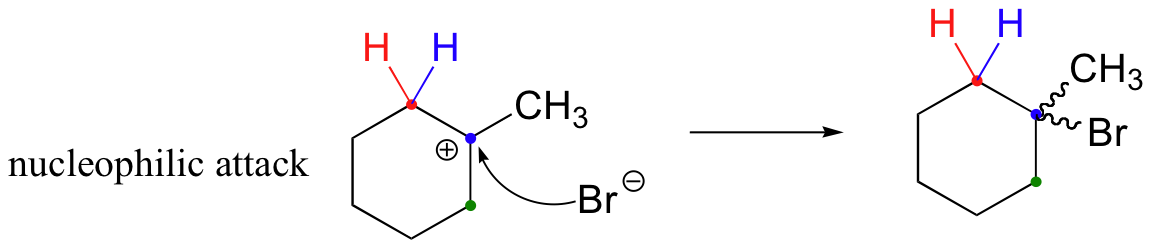
The first 1,2-alkyl shift is driven by the expansion of a five-membered ring to a six-membered ring, which has slightly less ring strain. A hydride shift then converts a secondary carbocation to a tertiary carbocation.
Carbocation rearrangements are often problematic for synthetic organic chemists, as they can complicate what at first glance appears to be a straightforward transformation. Directly introducing an n-propyl substituent to benzene in a Friedel-Crafts alkylation reaction (section 15.6), for example, is not feasible.
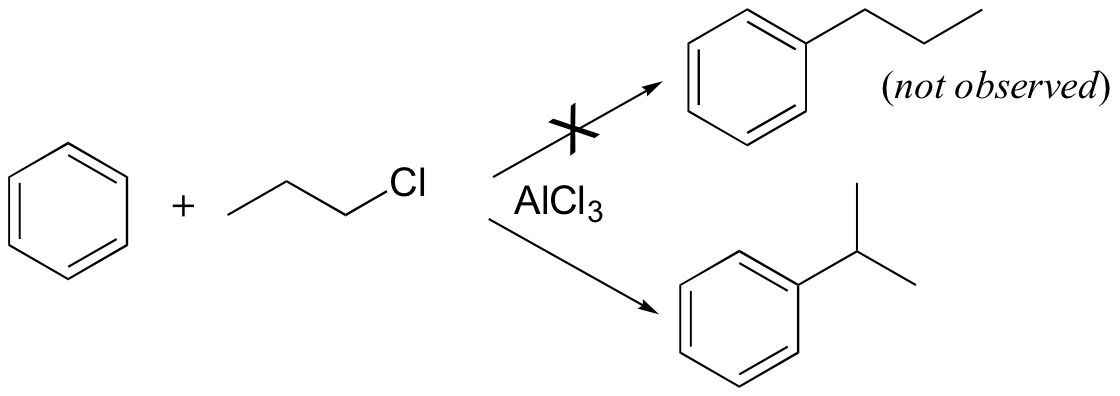
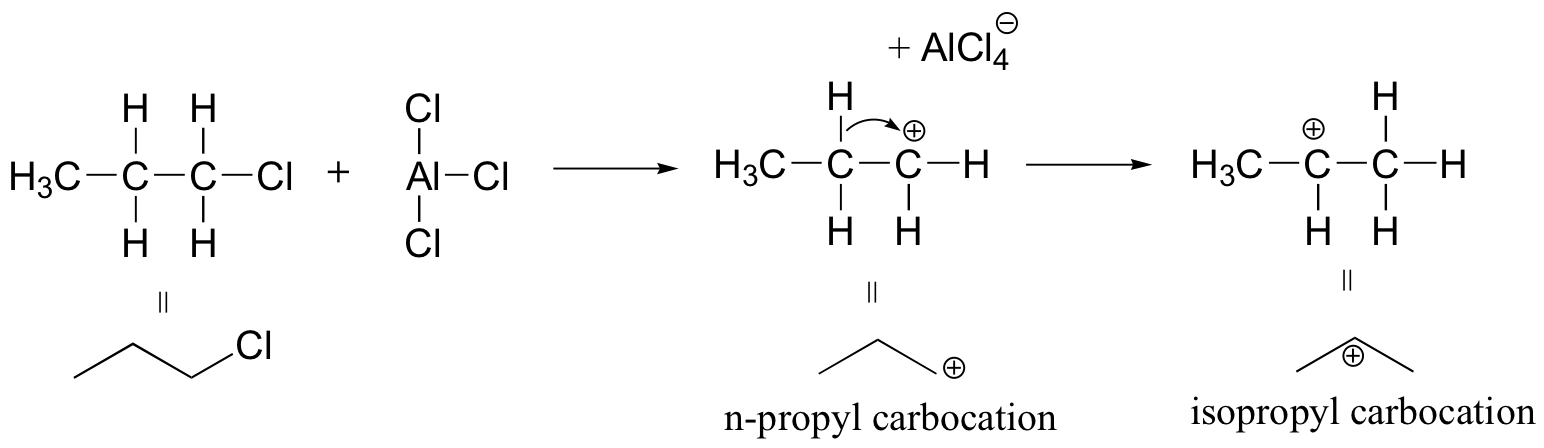
Because the adduct that forms between 1-chloropropane and AlCl3 reacts like a carbocation, a hydride shift rapidly occurs which results in the introduction of an isopropyl rather than an n-propyl electrophile.
In some cases, synthetic chemists have developed ingenious methods to get around the possibility of carbocation shifts. Suppose, for example, that we wanted to carry out the following alkene hydration:
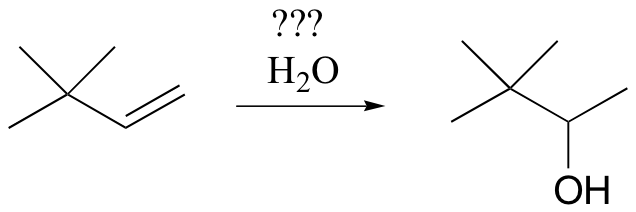
You might think that we could simply use aqueous acid. The problem with this strategy becomes evident, however, when the secondary carbocation intermediate is formed – this intermediate is likely to undergo a methyl shift to form a the more stable tertiary carbocation, which leads to a different product that the one we are aiming for.
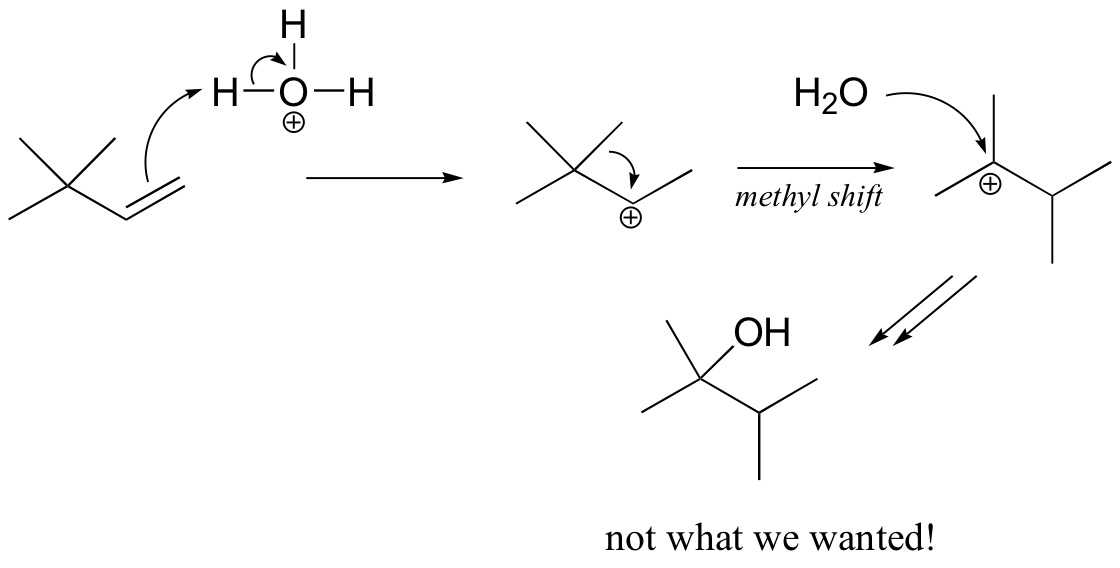
The solution in this case is to use a set of reaction conditions known as oxymercuration-reduction. Notice here that, unlike in the simple acid hydration, rearrangement does not occur.
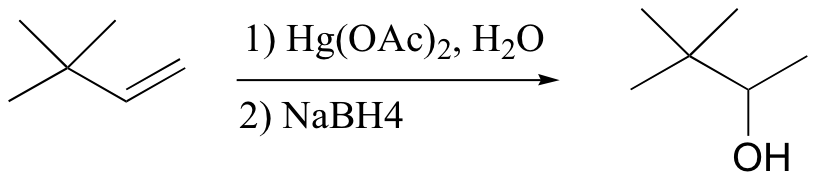
How does oxymercuration-reduction work? First, the mercury acetate Hg(OAc)2 dissociates to form a positively charged mercury acetate electrophile.

When the p electrons of the alkene attack the mercury cation, a three-membered intermediate forms in which both carbons share a single bond with the mercury and a partial positive charge. It turns out that rearrangements are unlikely to occur from this intermediate (as opposed to the discreet carbocation intermediate in the acidic hydration reaction).
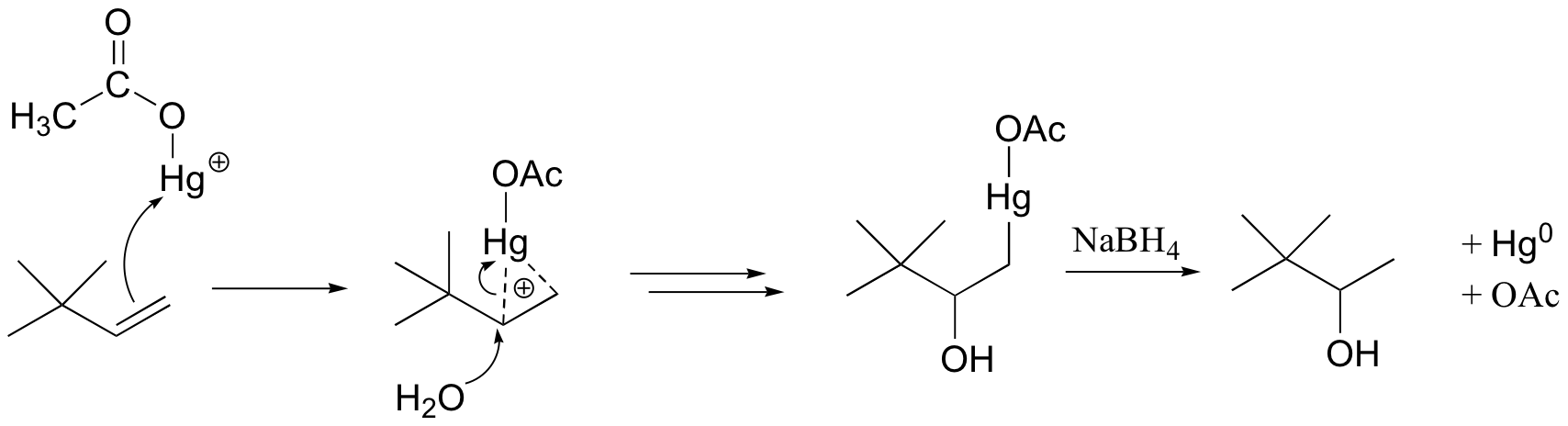
A water molecule then attacks the more substituted carbon, which bears a greater share of the positive charge. Cleavage of the carbon-Hg bond is accomplished with a reducing agent called sodium borohydride (NaBH4). We will not worry about the mechanism of this step at this stage, but it will make a lot more sense after we cover sodium borohydride reduction reactions in section 16.13.
The end result of an oxymercuration-reduction reaction is the Markovnikov addition of water to an alkene without rearrangement.
15.7B: Enzymatic reactions with carbocation rearrangement steps
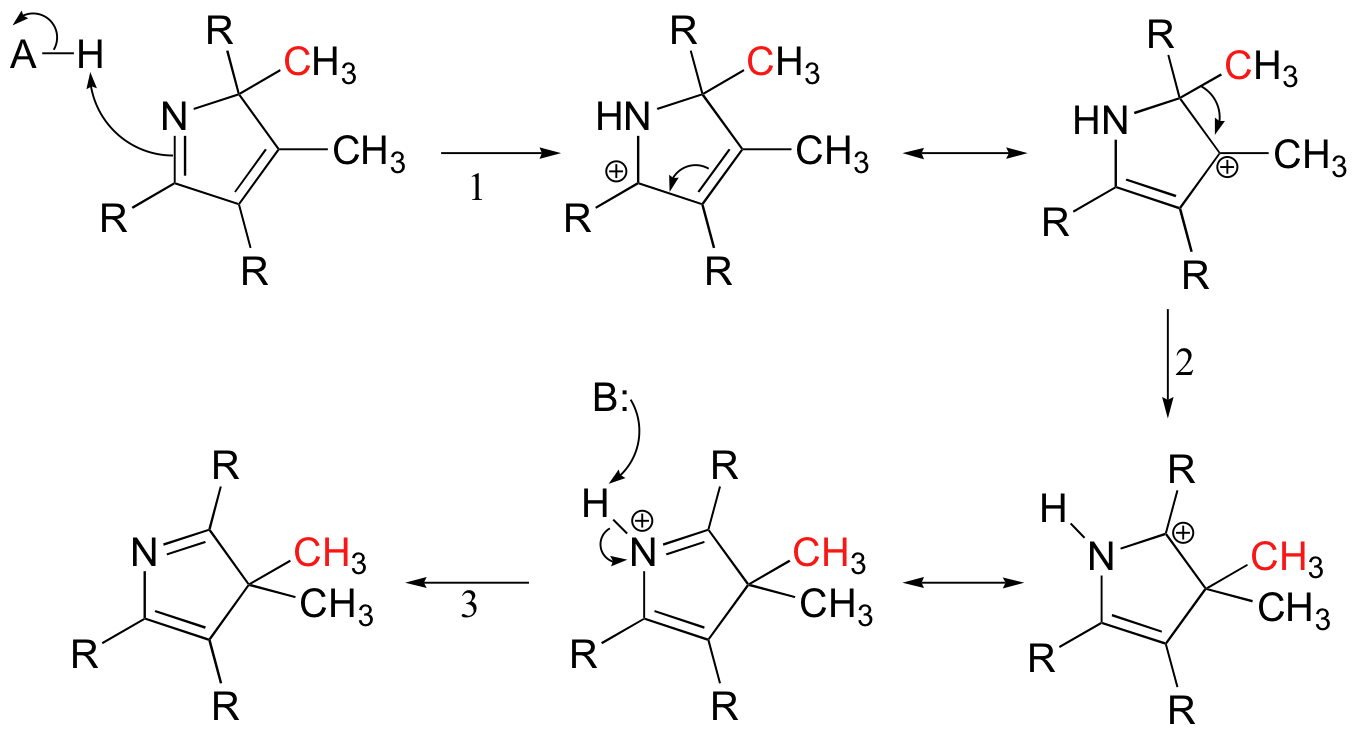
Carbocation rearrangements are involved in many known biochemical reactions. The biosynthesis of vitamin B12, for example, includes a reaction with a 1,2-methyl shift in step 2:
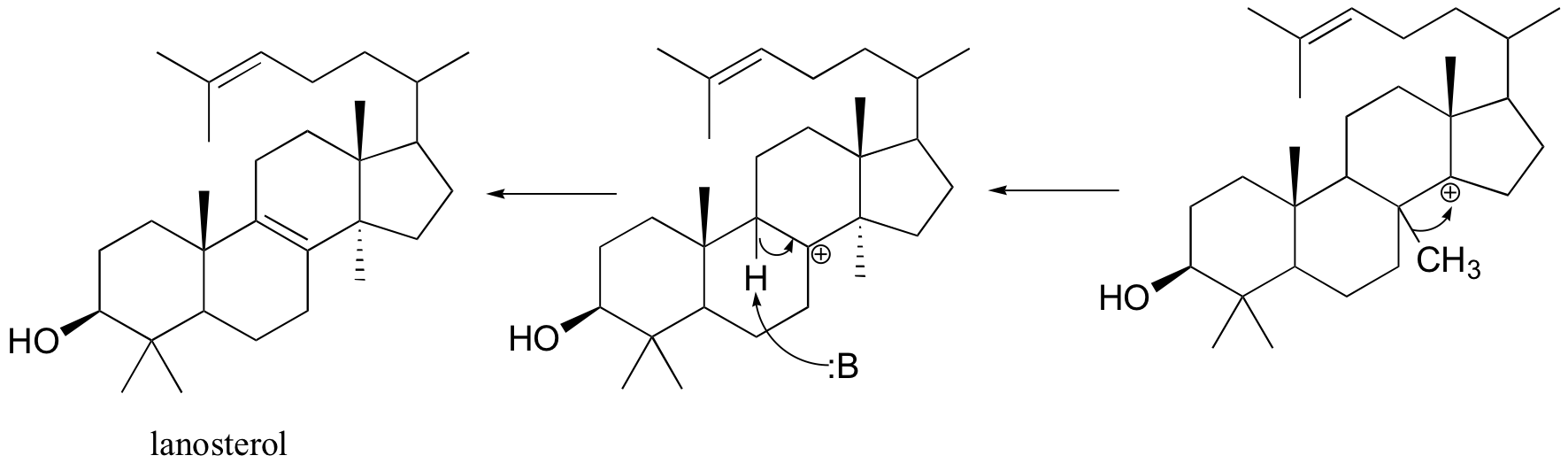
Rearrangements are particularly important in carbocation-intermediate reactions in which isoprenoid molecules cyclize to form complex multi-ring structures. One of the key steps in the biosynthesis of cholesterol is the electrophilic cyclization of oxidosqualene to form a steroid called lanosterol.
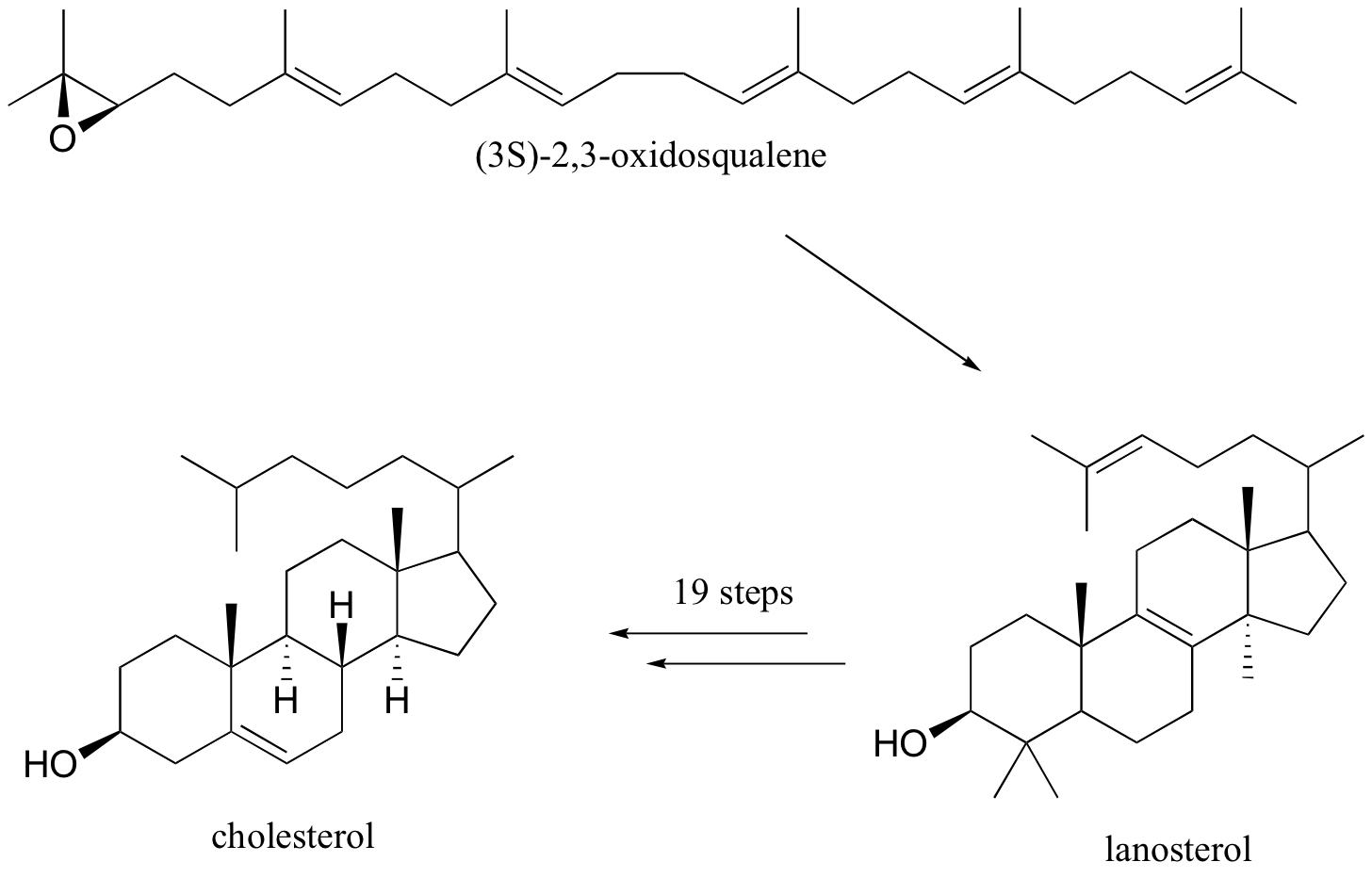
This fascinating reaction has two phases. The first phase, in which the actual cyclization takes place, is a series of electrophilic addition steps. The second phase is a series of hydride and methyl shifts. There is some argument about whether these processes occur in a stepwise fashion (with discreet carbocation intermediates) or in a concerted manner. For the sake of clarity, we will show the reaction proceeding stepwise.
The cyclization phase begins with attack by pi electrons on an epoxide electrophile (step 1 - review epoxide ring-opening reactions in section 8.6B).
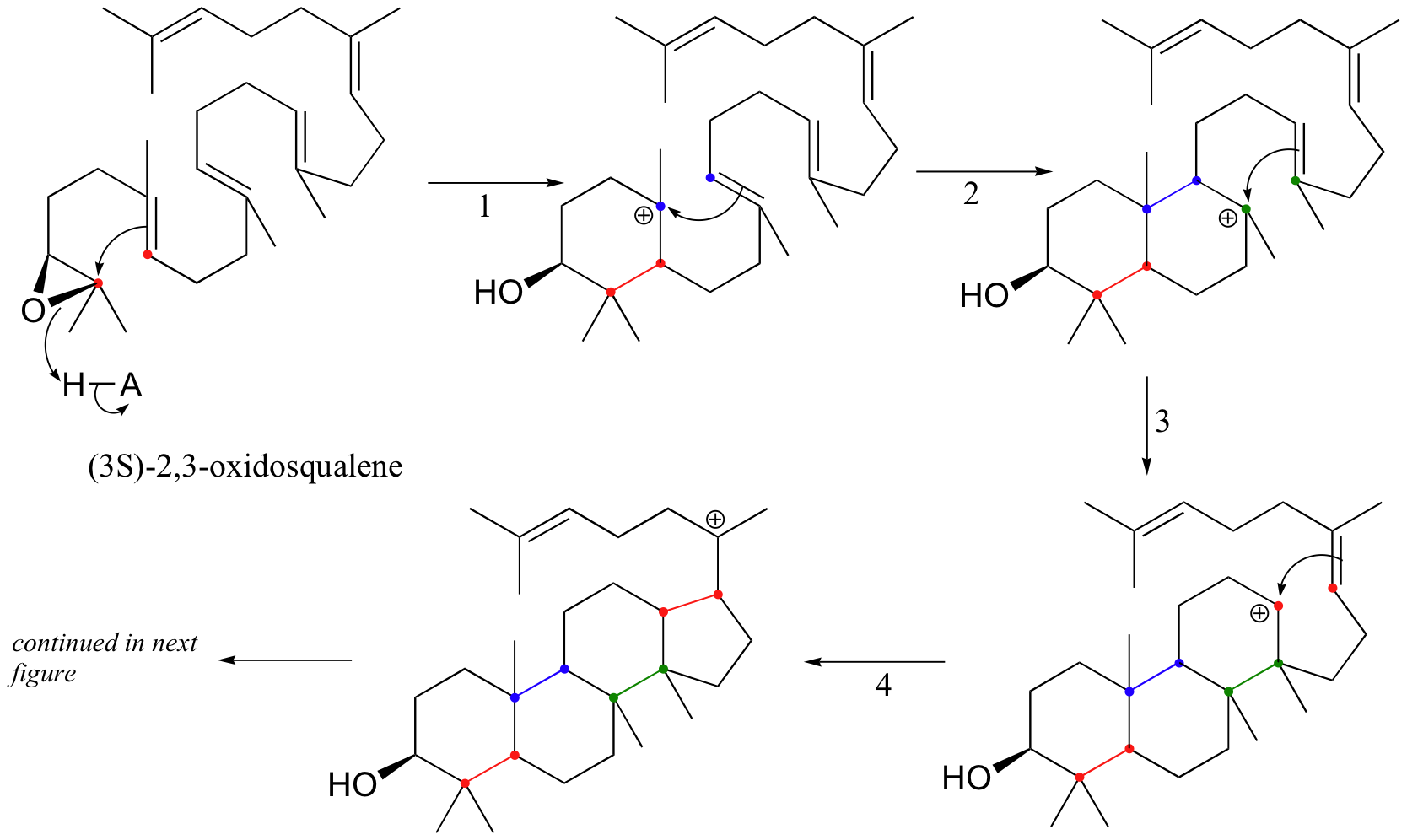
Steps 2, 3, and 4 are simply successive attacks by pi electrons on the carbocation generated by the previous attack. The overall result of this electrophilic cascade is the opening of the epoxide ring, and closure of three six-membered and one five-membered ring.
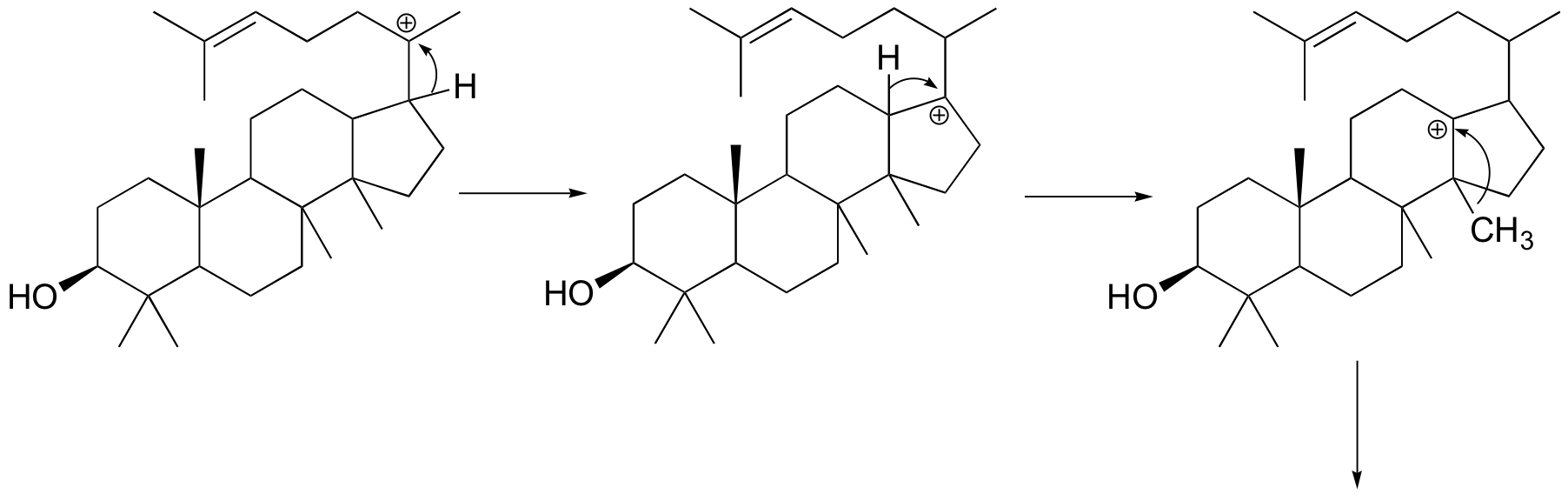
Next comes the rearrangement phase of the reaction. which is a series of two hydride shifts and two methyl shifts, followed by a proton abstraction which finally quenches the positive charge to form lanosterol.
Another well-studied isoprenoid cyclization is the cyclization of the 15-carbon isoprenoid farnesyl diphosphate to form pentalenene (Science 1997, 277, 1820). Pentalenene is a key precursor to a family of antibiotic compounds made by Streptomyces bacteria.
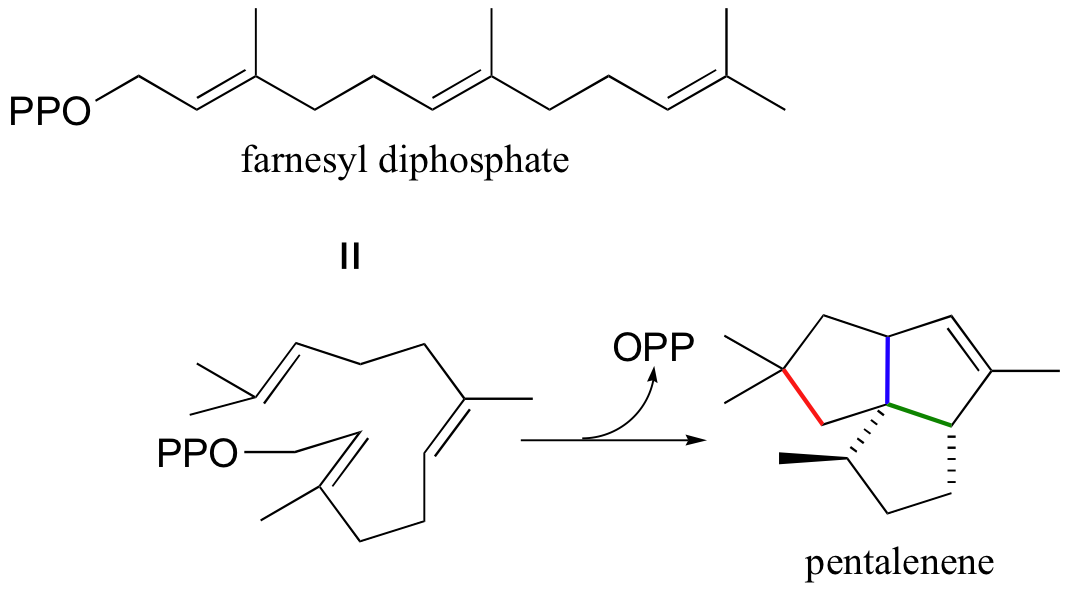
The cyclization process begins with the departure of the pyrophosphate leaving group and formation of an allylic carbocation. After three pi bond attacks, two hydride shifts, and a deprotonation (not in that order), the triple fused-ring structure of pentalenene is formed. The acid-base group in this reaction is an active site histidine.

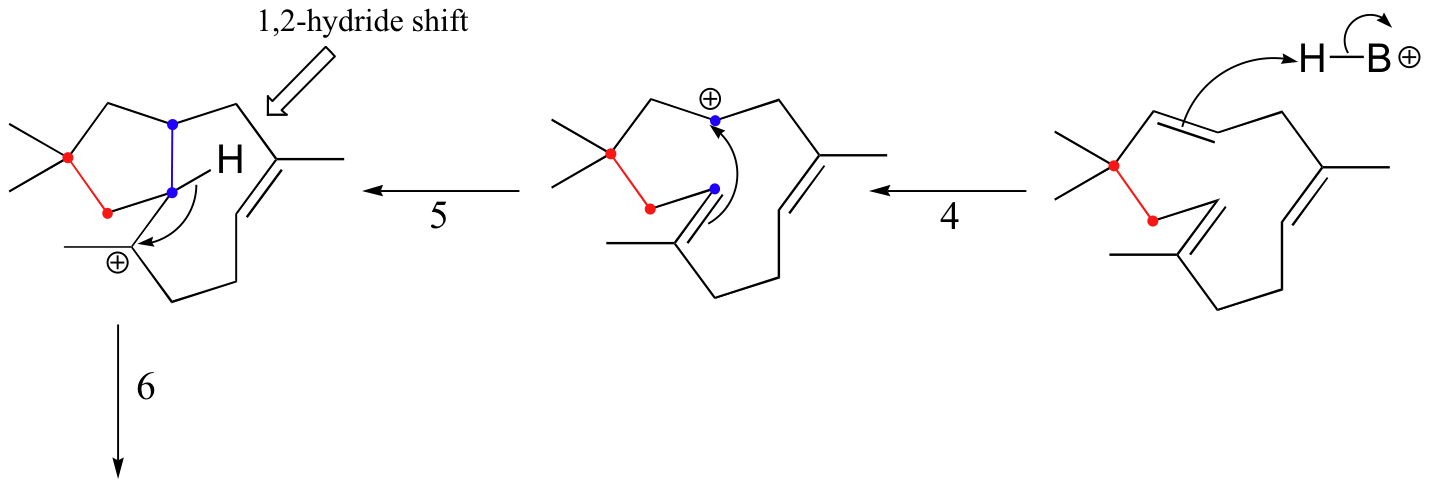
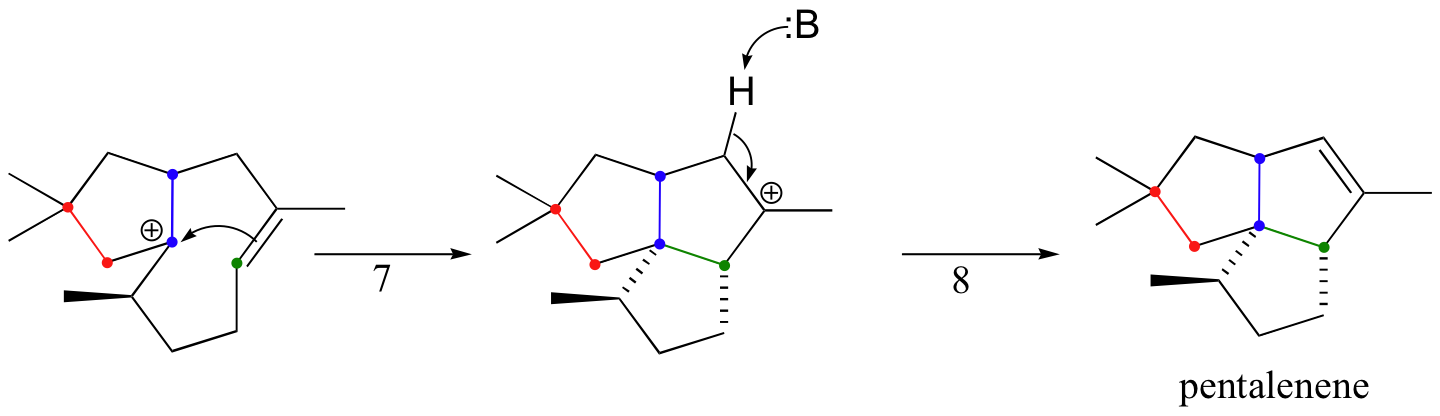
Notice that steps 3 and 4, which are enzyme-mediated deprotonation/protonation steps, essentially accomplish the same thing as a 1,2 hydride shift. A 'real' hydride shift, which is accomplished without the assistance of an acid-base enzyme group, is seen in step 6.
These isoprenoid cyclization reactions are truly remarkable examples of the exquisite control exerted by enzymes over the course of a chemical reaction. Consider the pentalenene synthase reaction: here, a relatively simple starting molecule without any stereocenters is converted, by a single enzyme, into a complex fused-ring structure with four stereocenters. The starting compound, farnesyl diphosphate, could potentially cyclize in many different ways, resulting in a great variety of different products - indeed, several hundred products of farnesyl diphosphate cyclization have been characterized from various natural sources (there are a couple of examples of these in the problem set for this chapter). In order for pentalanene synthase to catalyze the formation of a single product with the correct connectivity and stereochemistry, the enzyme must be able to precisely control the active site conformation of the starting compound and all reactive intermediates - otherwise, the wrong product will form.
15.7C: The acyloin, pinacol, and Hoffman rearrangements
Hydride and alkyl rearrangements can also start from a position adjacent to a carbonyl group. In an acyloin rearrangement, an alkyl group on an alpha-hydroxy carbonyl compound migrates directly to the carbonyl carbon:
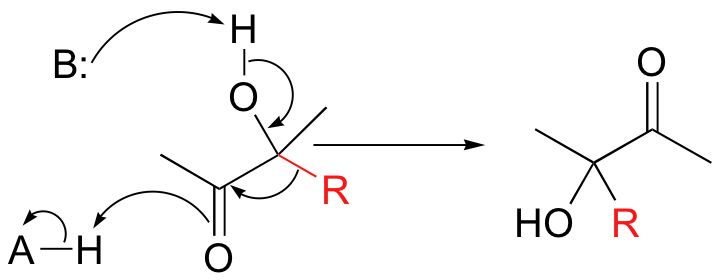
When vicinal diols are treated with sulfuric acid, the pinacol rearrangement is observed (a vicinal diol is a compound with two hydroxyl groups on adjacent carbons):
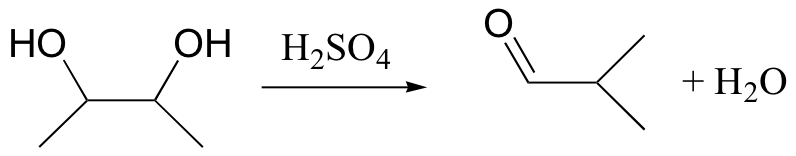
You will have a chance to draw a mechanism for this rearrangement in the end-of-chapter problems.
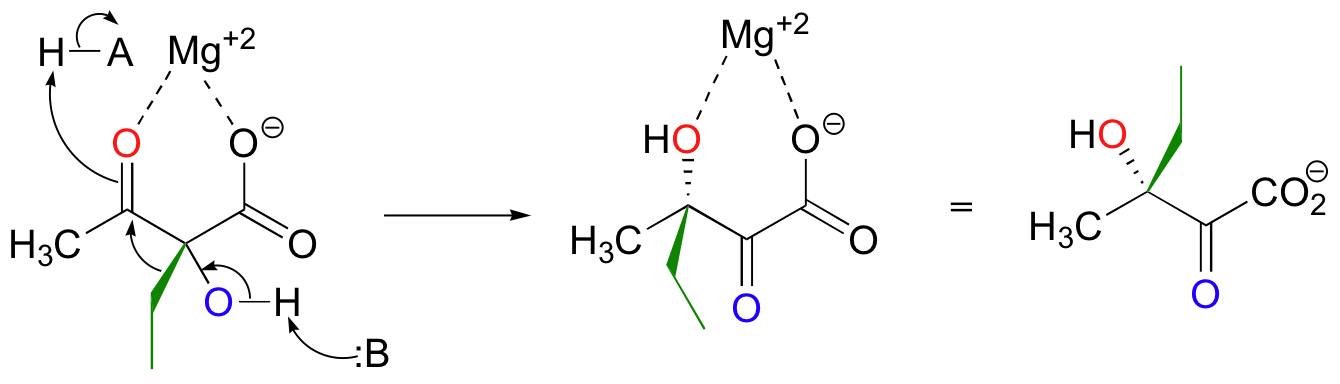
Both the acyloin and the pinacol rearrangements are well-known laboratory reactions, but enzymatic examples can also be found. This interesting acyloin rearrangement step, for example, occurs as part of the biosynthesis of the branched-chain amino acid isoleucine.
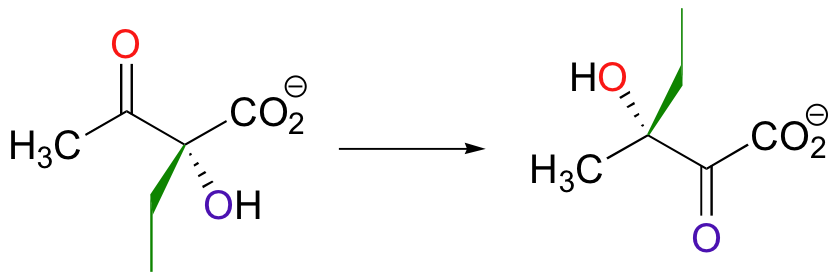
Magnesium ion coordination to the ketone oxygen increases the partial positive charge on C2 of the starting compound. The ethyl shift then shifts from C3 to C2, assisted by a 'push' from the deprotonation of the C3 oxygen and a 'pull' from the protonation of the C2 oxygen.
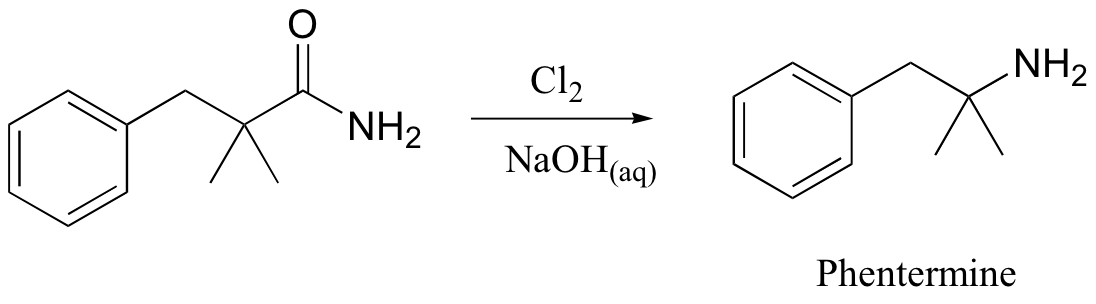
A somewhat complex but synthetically very useful rearrangement is known as the Hoffman rearrangement, or the Hoffman amide degradation. Essentially, an amide (with no N-substitution) is converted to the corresponding amine:
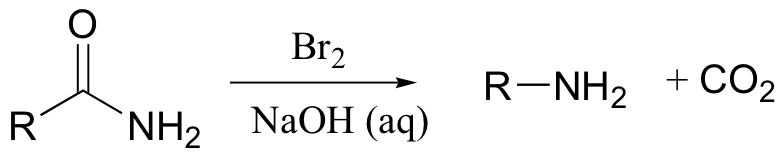
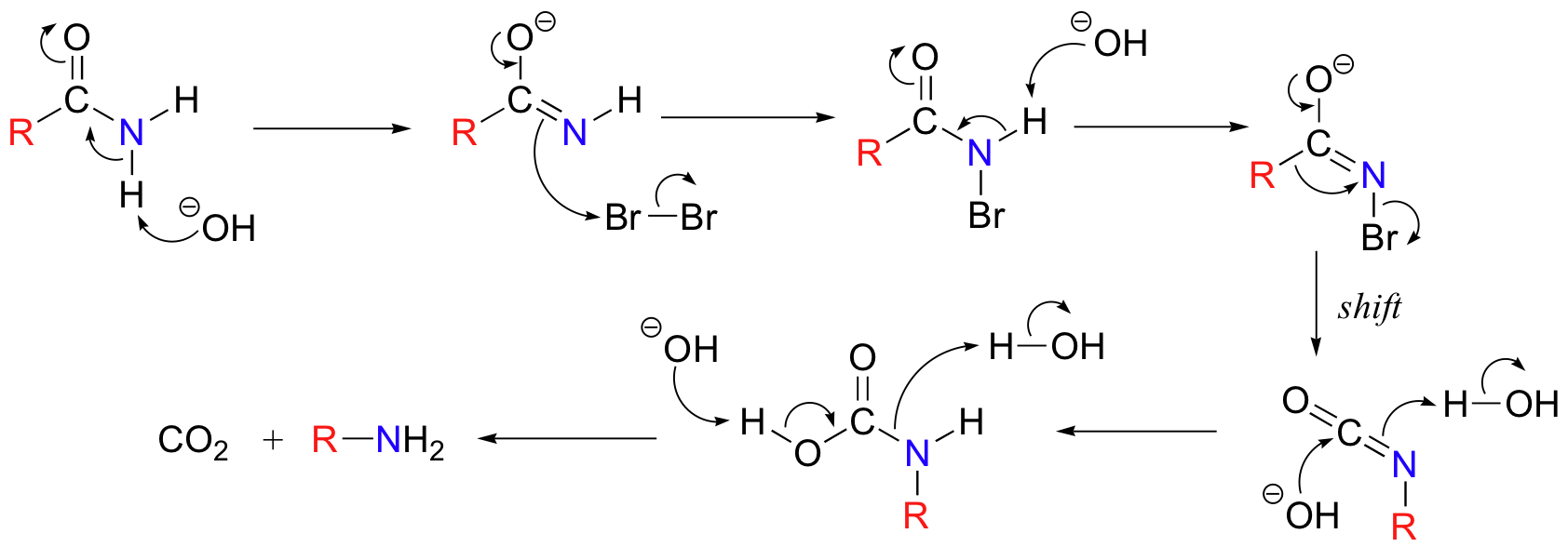
Below is the mechanism. You need not worry about learning this mechanism by heart, but you should be able to follow through each of the steps and recognize that each one makes chemical 'sense'. Notice the alkyl shift that occurs in the fourth step, and that the final step is a decarboxylation.
As stated above, this is in fact a useful reaction in the synthesis lab. The diet drug phentermine is synthesized via a Hoffman rearrangement (phentermine is one-half of the drug combination commonly known as Fen-Phen, which was taken off the market in 1997 due to its association with heart damage).