
12: Woodward’s Synthesis of Chlorophyll
- Page ID
- 21851

Chlorophyll, the most conspicuous of natural products, has held the fancy of organic chemists for more than two century, not only for its complex structure but also for its photochemical roll in the production of food that sustains all living things on this planet. Isolation and structure elucidation work began during the end of nineteenth century. This monumental activity that started with Willstätter, culminated in elucidation of the complete structure for Chlorophyll ‘a’ only by the middle of twentieth century. Most of these outstanding works, stretched over half a century, took place when modern organic chemistry was at its infancy. The synthesis of Chlorophyll ‘a’ by R. B. Woodward1 is acclaimed was an outstanding achievement in organic synthesis and ranks amongst the shinning gems in synthesis. The preliminary analysis for the synthesis, the persistent planned attach on this complex, delicate chemistry by his school and the logic of the famous Woodwardian approach are all good lessons for any discerning student of Organic Synthesis.
Analysis before initiating the synthesis
In the cited lecture, Woodward starts his discussions on synthesis with a series of questions that delineates not only the critical features of the structure under attach, but also arrives at a plausible target and approach for synthesis of this complex molecule. The structure of Chlorophyll ‘α’ is shown in (Fig 12.1).
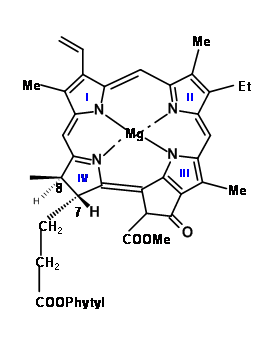
Earlier work had established that the magnesium atom and the phytyl group were easy to remove and put in place through well established chemistry. Chlorophyll ‘α’ belongs to the green coloured pigment family chlorins (12.2). Simple chlorins are readily oxidized (lost their ‘extra’ hydrogens) to give the more conjugated porphyrins (12.3). But some chlorins like chlorophyll do so only under drastic conditions. The second feature that drew their attention is the carbocyclic five membered fused ring attached to ring III. A close study of the molecular models suggested that a porphyrin, which had a string of substituents on C5, C6, Cγ, C7 and C8 positions, would be highly crowded as shown in (12.3) and (12.4). They noted that chlorins and porphyrins that had a substitution at Cγ and a carboxyl group at C6, loose CO2 with ease, while absence of Cγ subsitution
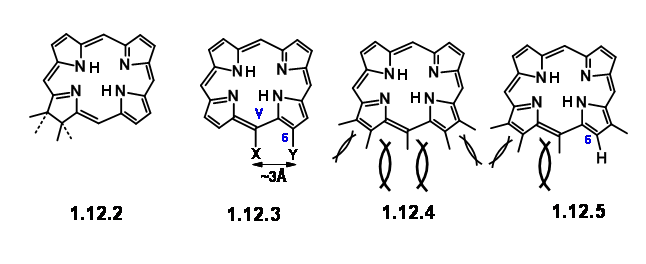
endeared the C6 carbonyl increased stability. All these features led them to conclude that in porphyrins, the space at C7 / Cγ and Cγ / C6 are most crowded. Decarboxylation reduces this strain partially(12.5). Presence of substituent at Cγ increases the strain for C7 / Cγ space as well. This crowding is perhaps partly relieved by formation of the carbocyclic ring at Cγ / C6. Furthermore, the strain at C7 / Cγ could be relieved by introduction of the ‘extra’ hydrogen at C7 and C8. The same strain factors would influence the trans- arrangement for substituents at C7 and C8. Such detailed analysis of the given structure not only helped them to understand the given structure but also suggested that the ‘Target’ for synthesis could be a porphylin like (12.6). Once it is synthesized, such a structure could be coerced to move on to take up the ‘extra’ hydrogens and move to (12.7). Some questions remained. The vinyl group on the first ring was considered as very sensitive to withstand the rigour of the projected synthesis.
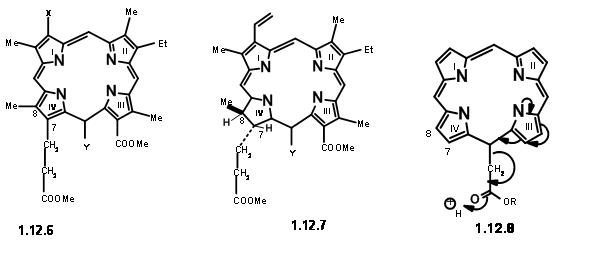
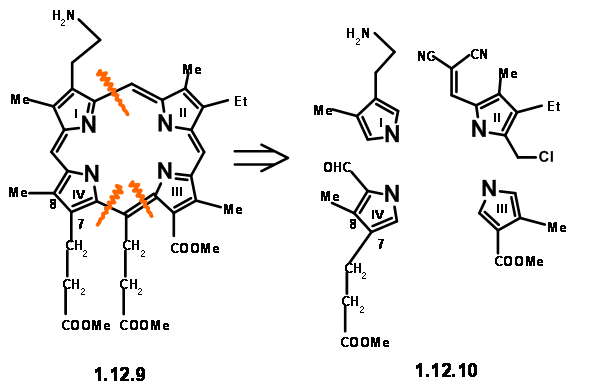
Hence, it was replaced with an equivalent aminopropane chain. As shown in structure (12.7), the residue needed at Cγ is an acetic acid moiety. However, mechanistic analysis of such a porphyrin (12.8)suggested that such a unit would readily eliminate due to an expected ‘electronic factor’ (actually, we would now say ‘enamine-like activity’). They decided to make this unit a propionic acid residue to avoid this instability. We have now arrived at a target structure (12.9). Based on the known pyrrole chemistry, they decided to make the Right Hand Side and the Left Hand Side of the molecule independently and condense these units into a tetramer. Thus, they arrived at the following four monomer units (12.10).
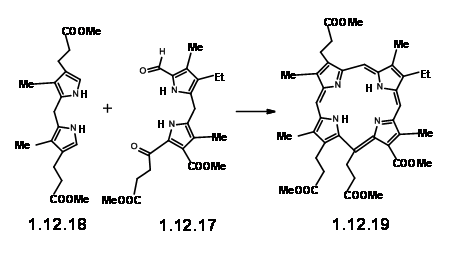
RHS Unit: The pyrrole units II and III were combined to give the expected dimer unit in good yields. Acylation with β-carbomethoxypropionyl chloride gave the RHS unit (12.17).

With this RHS unit (12.17) on hand, they tried the crucial cyclisation with a readily available model LHS unit (12.18). On condensation under acid conditions, followed by oxidation with iodine, the required porphyrin (12.19) could be obtained in acceptable 25% yield.
Encouraged with this result, they went ahead to synthesize the actual LHS unit (12.26).
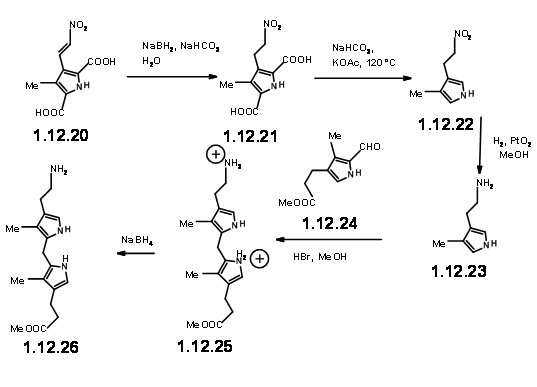
With the LHS unit ready at hand, they looked at the cyclisation of these two units. They realized that this condensation could give two products due to two different orientations of the reactants. Though the yield of this condensation product was comparable to previous condensation, they saw some drawbacks.
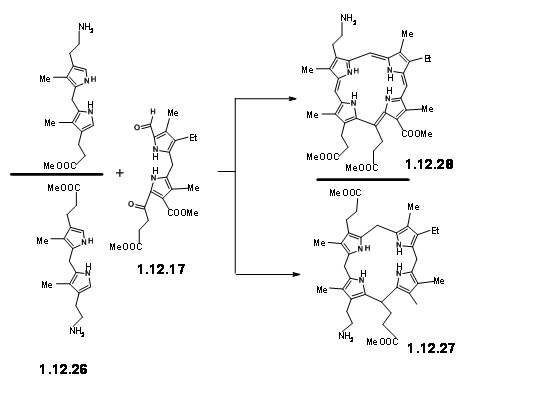
The yield was not acceptable for their projected purpose. Furthermore, this route resulted in the formation of isomeric products, whose structure assignment posed problems. Such ‘inelegance’ (an expression used by RBW in his lecture) was not acceptable for their group. Hence, they decided to freeze the mobility of the two units by linking the amine and the aldehyde units into a Schiff base (12.29).
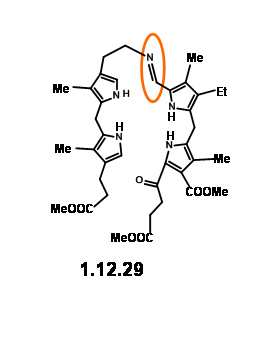
This masterly defined craftsmanship in molecular engineering was, however, not easy to achieve in practice. Pyrrole aldehydes were generally unreactive and could be converted to Schiff bases only under acid catalysis or buffered conditions. The problem arose from the LHS unit that proved to be very sensitive to such reaction conditions. No trace of the expected condensation product was observed. After several experiments, activation at aldehyde unit via Schiff bases followed by condensation with LHS unit via base exchange became a viable approach. But the discovery that such Schiff bases could be
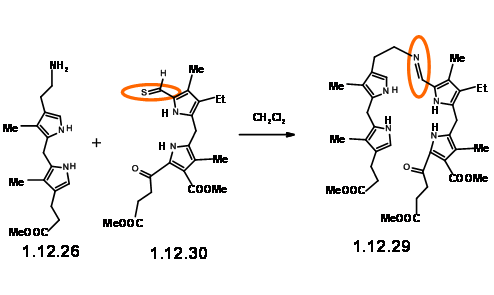
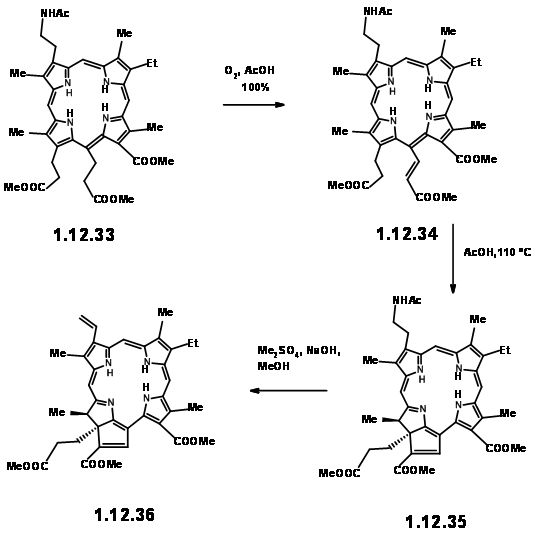
smoothly converted to thioaldehyde (12.30) provided a breakthrough. This thioaldehyde smoothly condensed with the LHS unit under neutral conditions in good yield. This ‘extraordinarily sensitive’ Schiff base, on acid treatment gave the cation (12.31), which was isolated as the dibromide. This dication was immediately oxidized with iodine to (12.32) and isolated as the acetamide (12.33) on acylation with acetic anhydride. In spite of the sensitivity of the intermediates and quick subsequent steps, the porphyrin (12.33) could be obtained in good yield and could be scaled up to several gram scale.

When the porphyrin (12.33) was heated in acetic acid in air, migration of the first hydrogen occurred to give (12.34). On heating in acetic acid at 110 0C, the chain at Cγ cyclised at C7 to give the reduced ring unit IV . Note the orientation of the two chains on ring IV is trans as expected. At this stage, attention was shifted to the vinyl group on ring I . This was readily achieved by Hoffmann exhaustive methylation-elimination sequence shown above (12.36).
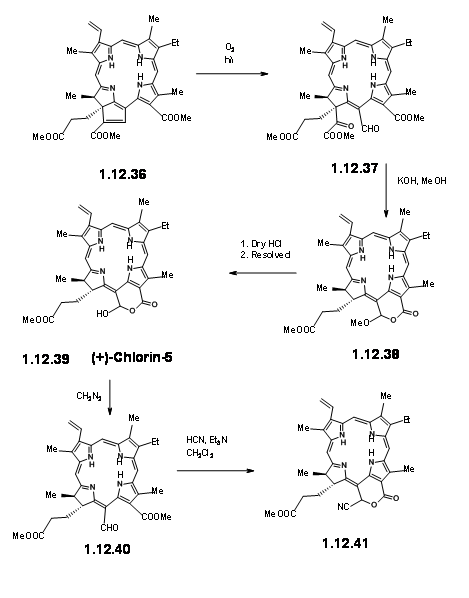
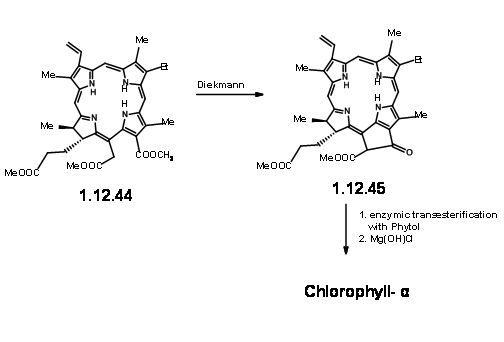
On exposure to light and air, the extra ring readily cleaved to give a pyruvic ester and a formyl group (12.37). The extra pyruvyl moiety was readily cleaved in methanolic KOH, which led to a methoxy lactone (12.380. Dry HCl gave the hamiacetal that could be resolved using quinine salt method to give (+)-Chlorin-5 (12.39). Diazomethane esterification exposed the aldehyde unit (12.40). Treatment of the formyl compound (12.40) with HCN in triethyl amine gave the cyanolactone (12.41), which on reduction, esterification and hydrolysis with methanolic HCl gave (12.44), the precursor for a Dieckmann cyclisation product.
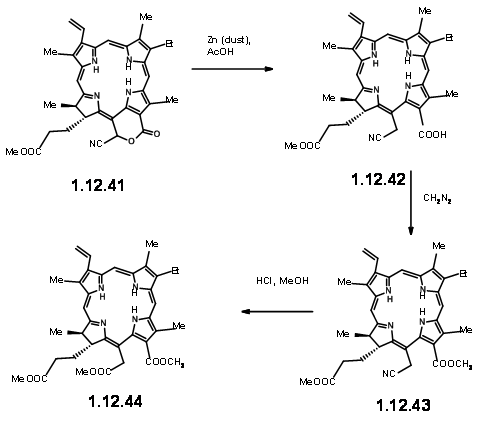
From here to (-)-Clorophyll α was already known. A Diekmann followed by introduction of the phytyl group by enzymztic methods completed the synthesis (Fortchr. Chem. Forsch., 2, 538 (1952)).
To this day, this masterpiece in organic synthesis has several feature that hold the interest for lovers of Art and Logic in organic synthesis.