8.1: Hydrosilylation of Alkenes
- Page ID
- 168815

Asymmetric hydrosilylation and hydroboration of carbon-carbon double bonds followed by oxidative cleavage of the C-Si and C-B bonds give effective methods for the construction of optically active alcohols (Scheme \(\PageIndex{1}\)).
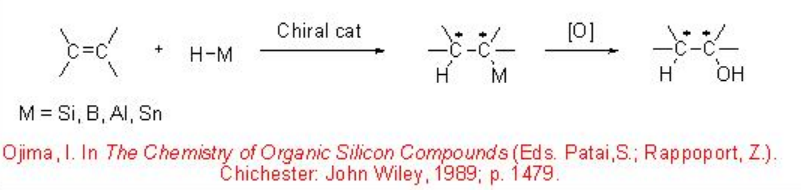
Asymmetric hydrosilylation of carbon-carbon unsaturated substrates provides effective methods for the synthesis of optically active organosilanes, which are versatile intermediates in organic synthesis. Chiral alkyl and aryl silanes can be converted into optically active alcohols with retention configuration by oxidative cleavage of a carbon-silicon bond into carbon-oxygen bond, while the diastereoselective reaction of chiral allyl- and allenyl silanes with C=O bond can give homoallylic and homopropargylic alcohols.
8.1.1 Reactions of Styrene and its Derivatives
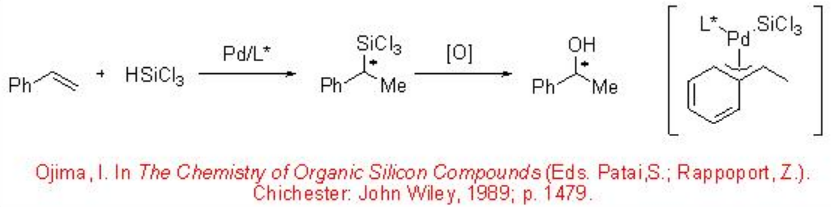
Chiral Palladium-catalyzed asymmetric hydrosilylation of styrene with trichlorosilane has been extensively studied. The reaction proceeds with excellent regioselectivity to give 1-phenyl-1-silylethane via a stable π -benzyl palladium intermediate (Scheme \(\PageIndex{2}\)).
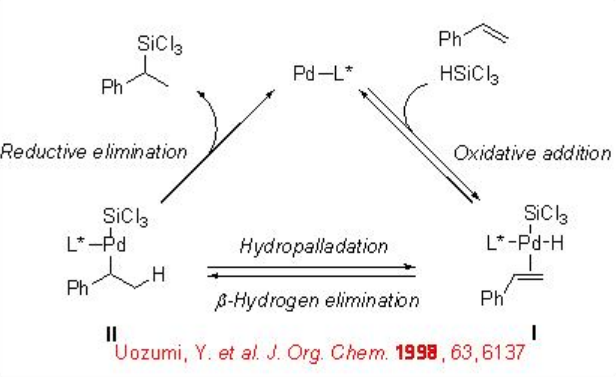
Scheme \(\PageIndex{3}\) illustrates the possible mechanism. Deuterium-labeling studies suggest that the β -hydrogen elimination is found to be much faster compared to the reductive elimination from the intermediate II . The involvement hydropalladation in the catalytic cycle has been revealed by the side product analysis from the reaction of o -allylstyrene.
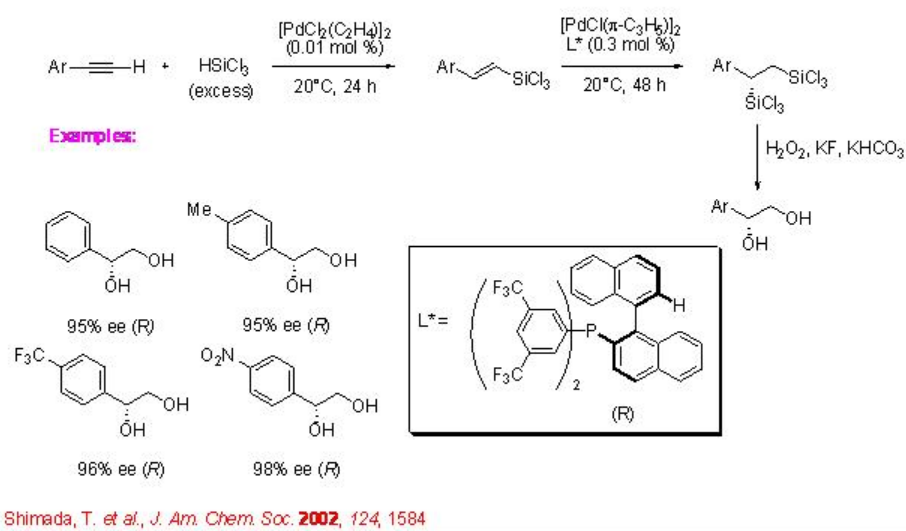
The reaction has been utilized in the synthesis of 1-aryl-1,2-diols from arylacetylenes (Scheme \(\PageIndex{4}\)). Platinum-catalyzed hydrosilylation of arylacetylene gives (E )-1-aryl-2-(trichlorosilyl)ethanes that could be further reacted with trichlorosilane in the presence of chiral palladium complex to afford optically active 1-aryl-1,2-bis(trichlorosilyl)ethanes. The latter could be transformed into optically active 1,2-diol via oxidative cleavage of the carbon-silicon bond into carbon oxygen bond.
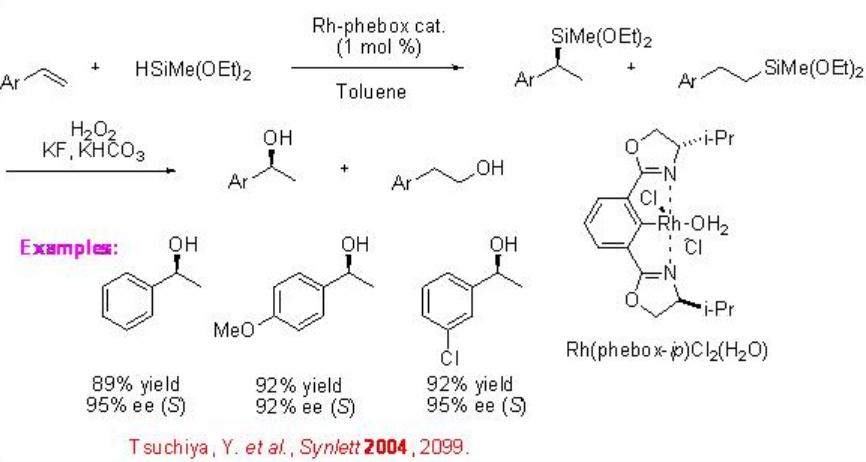
Other chiral catalysts have also been employed for the asymmetric hydrosilylation of alkenes. The chiral bis (oxazolinyl)phenylrhodium complex catalyzes the asymmetric hydrosilylation of styrenes with hydro(alkoxy)silanes in high enantioselectivity, although the regioselectivity is found to be somewhat moderate (Scheme \(\PageIndex{5}\)).
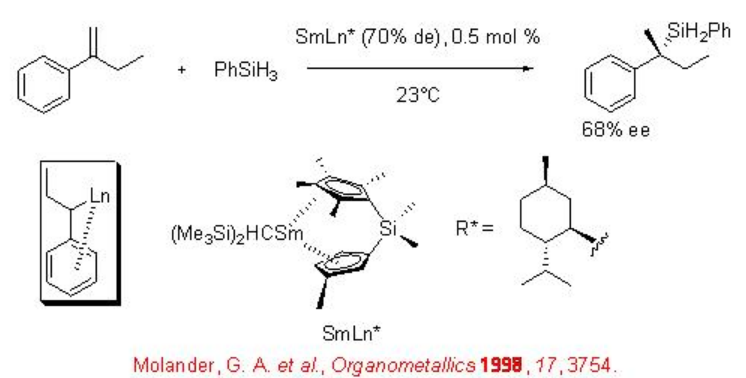
α -Substituted styrenes proceed reaction with phenylsilane to afford benzylic tert -alkylsilanes in the presence chiral organolanthanide as catalyst in moderate enantioselectivity (Scheme \(\PageIndex{6}\)).
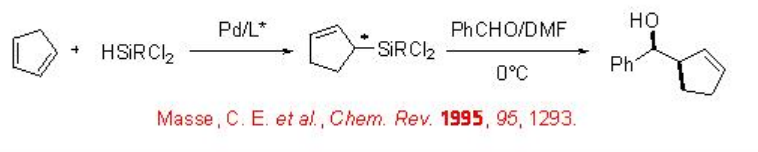
8.1.2 Reactions of 1,3-Dienes
The reaction of 1,3-dienes with hydrosilanes having electron-withdrawing groups on silicon affords synthetically useful optically active silanes in the presence of chiral palladium complex (Scheme \(\PageIndex{7}\)). The reaction proceeds in a 1,4-fashion providing chiral allylsilanes that could be converted into homoallylic alcohols on the reaction with aldehydes.
The use of ferrocenylphosphine and mop-phen ligands has been demonstrated for the hydrosilylation of cyclo-1,3-hexadiene in the presence of palladium salts (Scheme \(\PageIndex{8}\)). The reaction with phenyldifluorosilane afforded the highest enantioselectivity compared to that with trichlorsilane or methyldichlorosilane. Based on the reaction of with deuterium-labeled silane the involvement of π-allylpalladium intermediate and 1,4-cis-addition has been proposed.
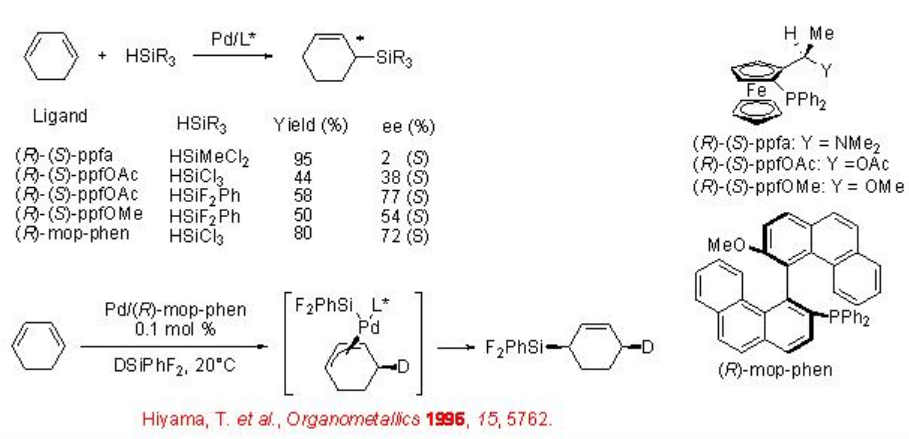
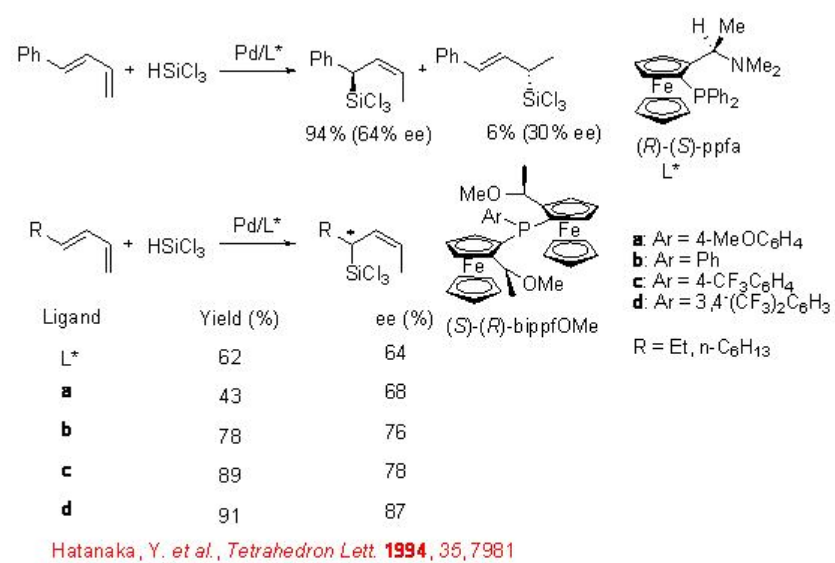
In case of linear 1,3-dienes, the regioselectivity has become an issue. In the reaction of 1-phenyl-1,3-butadiene using ferrocenyl ligand, (R)-(S)-ppfa, the formation of a mixture of regioisomeric allylsilanes is observed (Scheme \(\PageIndex{9}\)). However, in the reaction of alkyl substituted 1,3-dienes, 1,3-hexadiene and 1,3-decadiene, a single regioisomer is obtained with moderate enantioselectivity. Improvement in the enantioselectivity is observed employing the bis (ferrocenyl)monophophine ligands a-d having two planar chiral ferrocenyl moieties on phosphorus atom.
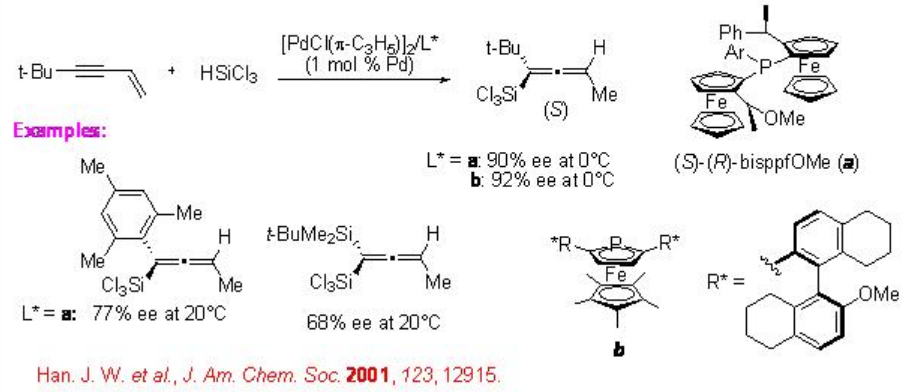
The reaction of 1-buten-3-ynes substituted with bulky groups at the alkyne terminus affords enantiomerically enriched allenylsilanes in the presence of palladium complex (Scheme \(\PageIndex{10}\)). For example, the reaction of 5,5-dimethyl-1-hexen-3-yne using (S)-(R)-bisppfOMe a proceeds in a 1,4-fashion to give allenyl(trichloro)silanes in high regio- and enantioselectivity. Further enhancement in the enantioselectivity is shown employing chiral phosphametallocene b having a sterically demanding η5 -C5 Me5 moiety.
8.1.3 Reactions of Alkyl Substituted Alkenes
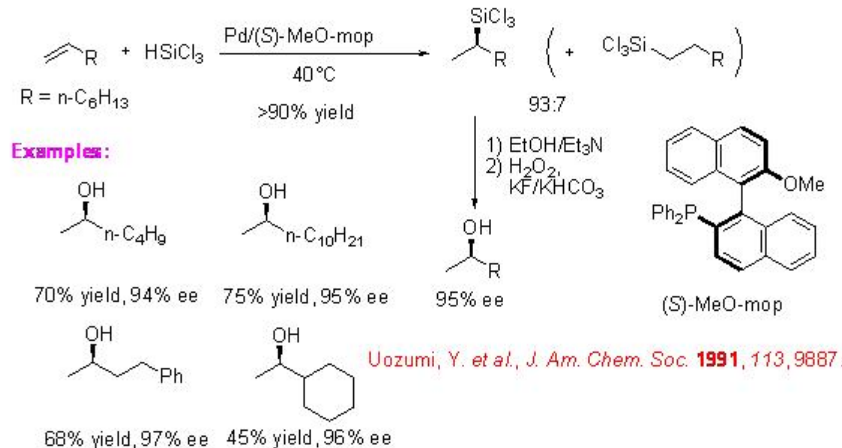
Hydrosilylation of simple terminal alkenes give branched products with high regioselectivity. The palladium systems show exceptional catalytic system compared to Pt, Ni and Rh based systems. For example, the hydrosilylation of 1-octene with trichlorosilane using palladium-(S)—MeO-mop gives a 93:7 mixture of 1-octylsilane and 2-octylsilane with 95% ee (Scheme \(\PageIndex{11}\)).
The above catalytic system is also effective for the hydrosilylation of cyclic alkenes, such as norbornene and bicyclo[2.2.2]octane, 2,5-dihydrofuran and norbornadiene. For example, the reaction of norbornene gives exo adduct exclusively (Scheme \(\PageIndex{12}\)). The hydrosilylated product can be transformed into exo -2-norbornanol or endo -2-bromonorbornane via the corresponding pentafluorosilicate. In addition, chiral ferrocenylmonophosphines a-d are too found to be effective for this process with excellent enantioselectivity.
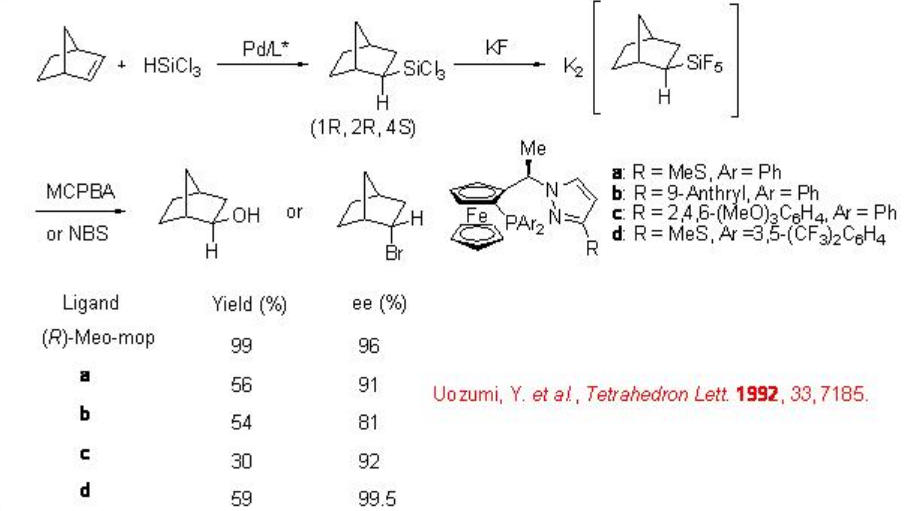
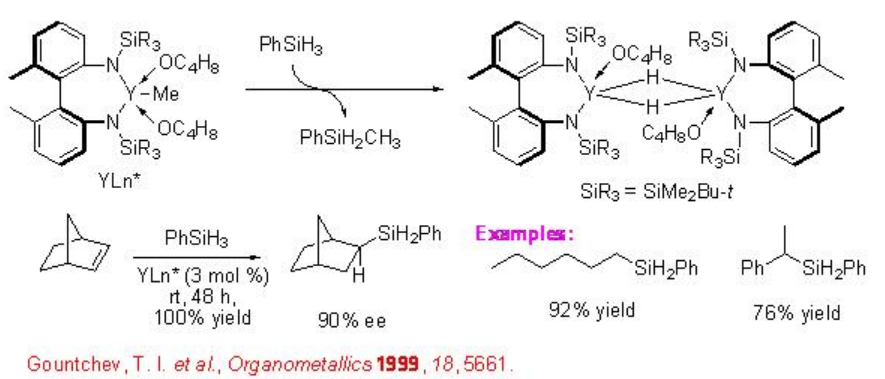
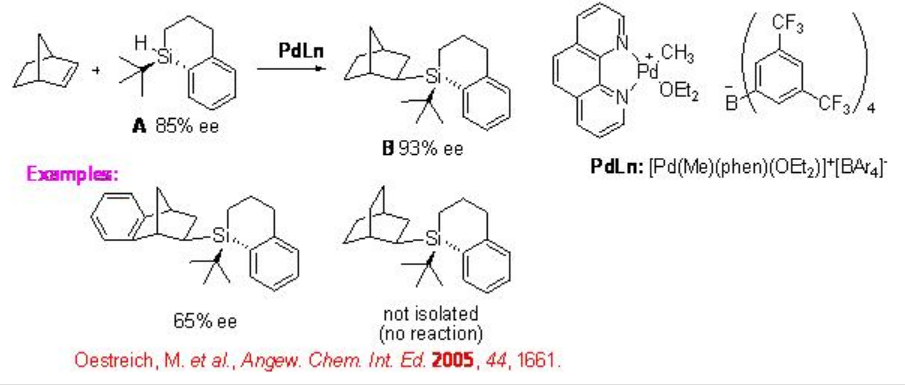
Chiral yttrium hydride complex (d0 metal complex) bearing non-Cp ligand catalyzes the hydrosilylation of norbornene with phenylsilane to produce exo -adduct with 90% ee (Scheme \(\PageIndex{13}\)). More recently, the first chirality transfer from silicon to carbon in a reagent-controlled reaction of norbornene is reported in the presence of achiral palladium complex. The hydrosilylation of norbornene with chiral silane A having 85% ee is found to form the hydrosilylated product B with 93% ee exhibiting asymmetric amplification (Scheme \(\PageIndex{14}\)).
8.1.4 Intramolecular Hydrosilylation
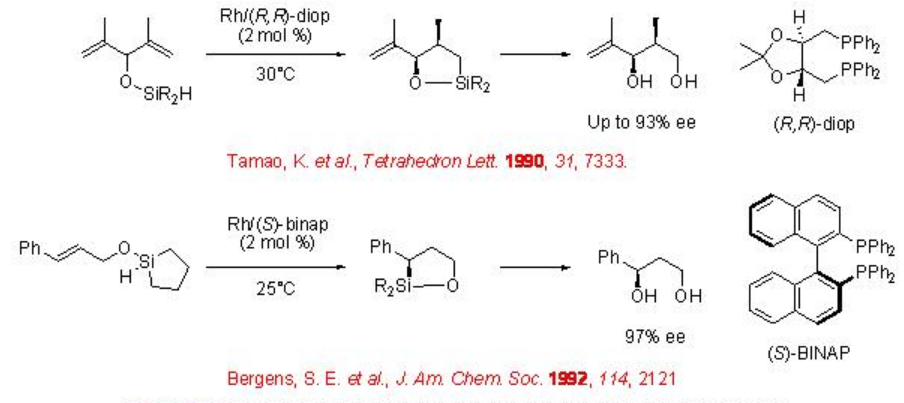
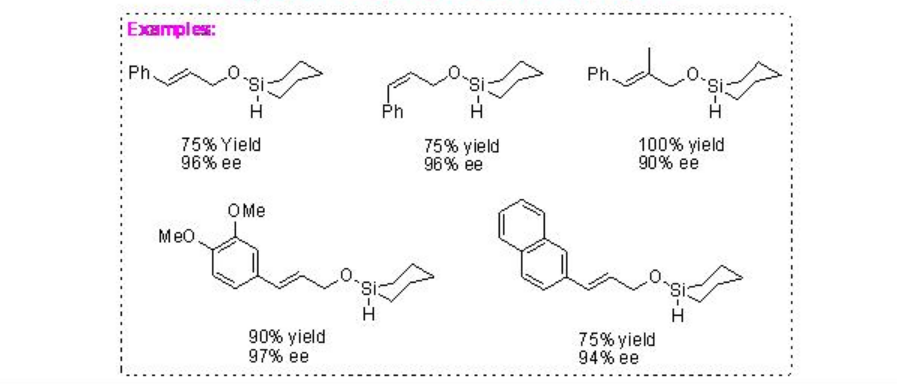
Synthesis of optically active polyols from allylic alcohols can be achieved using chiral Rh-catalyzed intramolecular hydrosilylation followed by oxidation of allyloxy hydrosilanes (Scheme \(\PageIndex{15}\)). For example, hydrosilyl ether of di(2-propenyl)methanol can be converted into optically active 1,3-diol using intramolecular hydrosilylation in the presence of chiral rhodium-(R,R)-diop followed by oxidation. Rh-BINAP is also found to be effective catalyst for the intramolecular hydrosilylation of hydrosilyl ethers of allyl alcohols.
8.1.5 Cyclization/Hydrosilylation
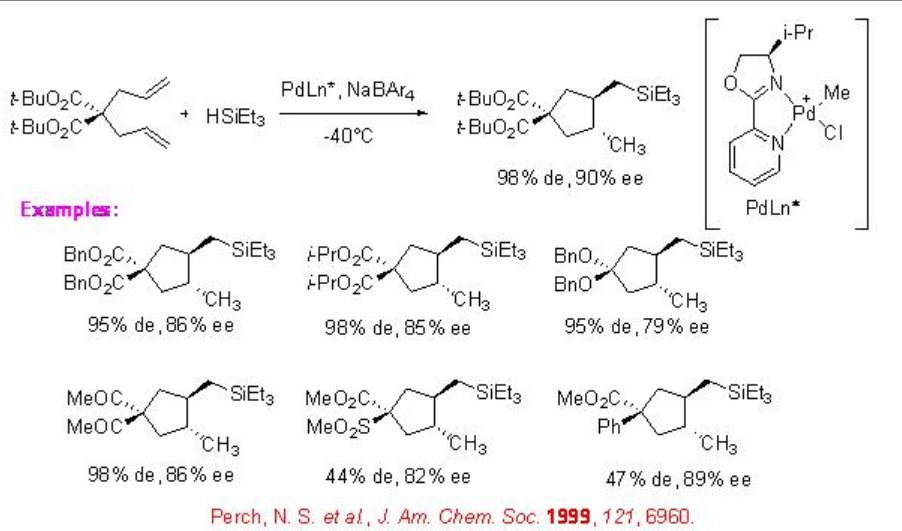
Asymmetric cyclization and hydrosilylation of a ,ω-diunsaturated compounds such as 1,6-dienes and 1,6-enynes affords powerful tool for the construction of optically active functionalized carbocycles. For example, the tandem reaction of diallylmalonate in the presence of cationic Pd complex bearing a chiral pyridine-oxazoline proceeds with high diastereoselectivity to yield the corresponding trans-substituted cyclopentane with 90% ee (Scheme \(\PageIndex{16}\)).
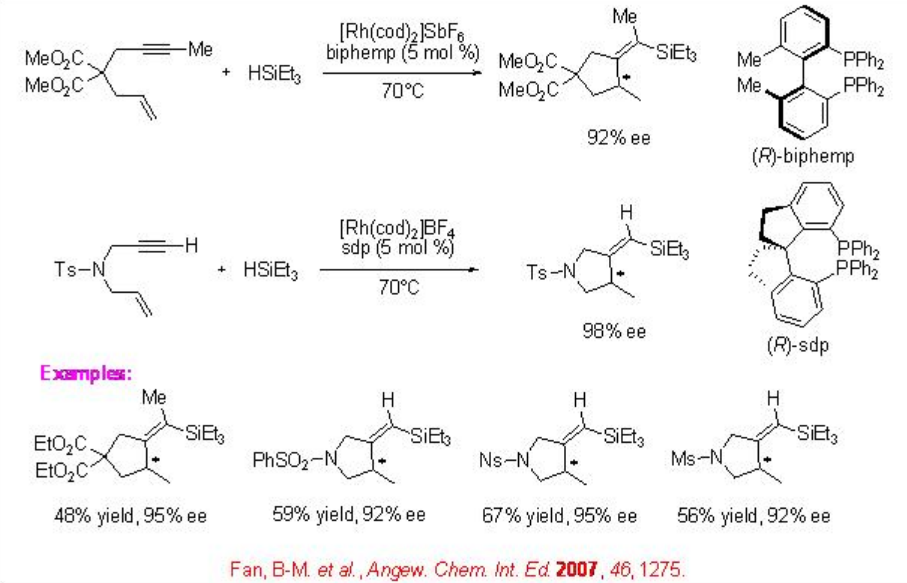
The reactions of 1,6-diynes using cationic Rh complexes bearing chiral bisphosphine gives the hydrosilylated alkylidenecyclopentanes with high enantioselectivity. For example, the 1,6-enyne proceeds reaction with triethylsilane in the presence of cationic Rh and ( R )-biphemp to give hydrosilylated alkylidene cyclopentane in 92% ee (Scheme \(\PageIndex{17}\)). Subsequently, chiral Rh complex containing spiro diphosphine ( R )-sdp is found to be effective for this process.
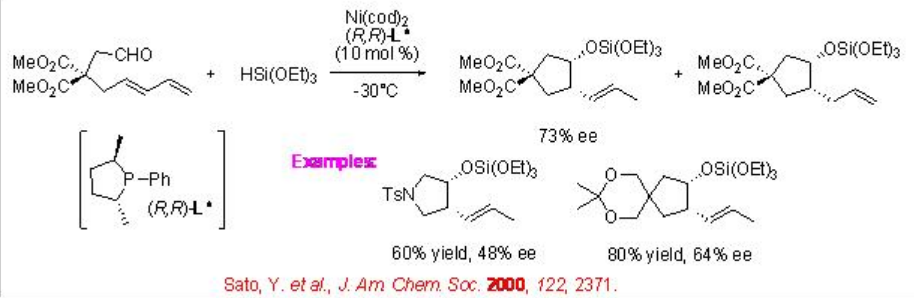
The synthesis of carbocycles can also be accomplished by the cyclization of ω-formyl-1,3-dienes in the presence of hydrosilanes and chiral nickel complex (Scheme \(\PageIndex{18}\)). For example, zerovalent nickel complex of (2 R ,5 R )-2,5-dimethyl-1-phenylphospholane catalyzes the cyclization of 1,3-dienes with a tethered formyl group in the presence of triethoxysilane to give five-membered carbocycle with 73% ee.