
26.4: Some Aromatic Side-Chain Compounds
- Page ID
- 22374

We have discussed in this chapter and in previous chapters how the reactivity of halogen, amino, and hydroxy substituents are modified when linked to aromatic carbons rather than to saturated carbons. Other substituents, particularly those linked to an aromatic ring through a carbon-carbon bond, also are influenced by the ring, although usually to a lesser degree. Examples include \(\ce{-CH_2OH}\), \(\ce{-CH_2OCH_3}\), \(\ce{-CH_2Cl}\), \(\ce{-CHO}\), \(\ce{-COCH_3}\), \(\ce{-CO_2H}\), and \(\ce{-CN}\), and we shall refer to aromatic compounds containing substituents of this type as aromatic side-chain compounds. Our interest in such compounds will be directed mainly to reactions at the side chain, with particular reference to the effect of the aromatic ring on reactivity. In this connection, we shall discuss the relatively stable triarylmethyl cations, anions, and radicals, as well as quantitative correlation of rates of organic reactions by what is known as the Hammett equation.
Preparation of Aromatic Side-Chain Compounds
Carboxylic acids can be obtained from most alkylbenzenes by oxidation of the side chain with reagents such as potassium permanganate, potassium dichromate, or nitric acid:

Under the conditions of oxidation, higher alkyl or alkenyl groups are degraded and ring substituents, other than halogen and nitro groups, often fail to survive. As an example, oxidation of 5-nitro-2-indanone with dilute nitric acid leads to 4-nitro-1,2-benzenedicarboxylic acid:
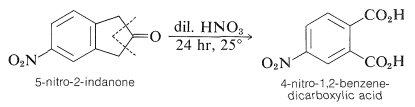
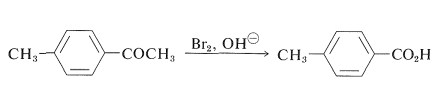
To retain a side-chain substituent, selective methods of oxidation are required. For example, 4-methylbenzoic acid can be prepared from 1-(4-methylphenyl)ethanone by the haloform reaction (Section 17-2B):
Many side-chain halogen compounds can be synthesized by reactions that also are applicable to alkyl halides (see table 14-5), but there are other methods especially useful for the preparation of arylmethyl halides. The most important of these are the chloromethylation of aromatic compounds (to be discussed later in this section) and radical halogenation of alkylbenzenes.
Light-induced, radical chlorination or bromination of alkylbenzenes with molecular chlorine or bromine was discussed previously (Section 14-3C). Under these conditions, methylbenzene reacts with chlorine to give successively phenylchloromethane, phenyldichloromethane, and phenyltrichloromethane:
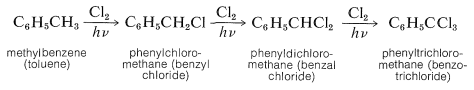
Related reactions occur with other reagents, notably sulfuryl chloride, \(\ce{SO_2Cl_2}\); tert-butyl hypochlorite, \(\ce{(CH_3)_3COCl}\); and \(\ce{N}\)-bromobutanimide, \(\ce{(CH_2CO)_2NBr}\). The \(\alpha\) substitution of alkylbenzenes is the result of radical-chain reactions (Section 14-3C).
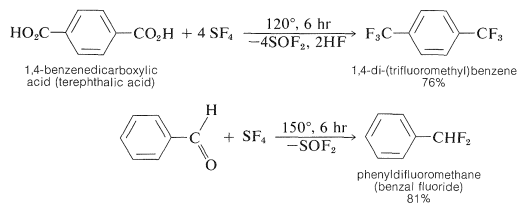
Side-chain fluorine compounds with the groupings \(\ce{-CHF_2}\), \(\ce{-CF_2}-\), and \(\ce{-CF_3}\) are available by the reaction of sulfur tetrafluoride or molybdenum hexafluoride with carbonyl compounds (see Section 16-4D):

Some specific examples follow:
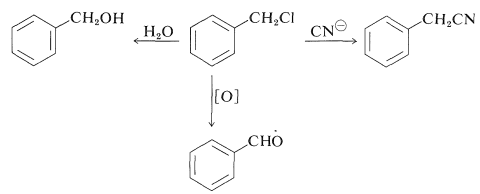
Arylmethyl chlorides or bromides are quite reactive compounds that are readily available or easily prepared, and as a result they are useful intermediates for the synthesis of other side-chain derivatives. Thus phenylmethyl chloride can be hydrolyzed to phenylmethanol, converted to phenylethanenitrile with alkali-metal cyanides, and oxidized to benzenecarbaldehyde (benzaldehyde):
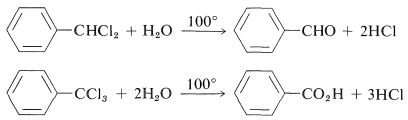
Phenyldichloromethane hydrolyzes readily to benzaldehyde, and phenyltrichloromethane to benzoic acid:
Carbon side chains also may be introduced by direct substitution and several such reactions have been discussed in detail previously. These include Friedel-Crafts alkylation and acylation (Section 22-4E and 22-4F), the Gattermann-Koch reaction for preparation of aldehydes from arenes and carbon monoxide (Section 22-4F), and the Kolbe-Schmitt, Reimer-Tiemann, and Gattermann reactions for synthesis of acids and aldehydes from arenols (Section 26-1E).
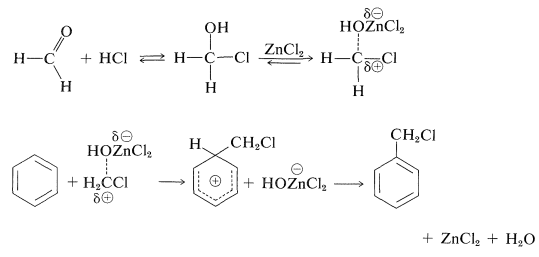
Chloromethylation is a useful method for substitution of \(\ce{-CH_2Cl}\) for an aromatic hydrogen, provided one starts with a reasonably reactive arene. The reagents are methanol and hydrogen chloride in the presence of zinc chloride:
\[\ce{C_6H_6} + \ce{CH_2O} + \ce{HCl} \overset{\ce{ZnCl_2}}{\longrightarrow} \ce{C_6H_5CH_2Cl} + \ce{H_2O}\]
The mechanism of the chloromethylation reaction is related to that of Friedel-Crafts alkylation and acylation and probably involves an incipient chloromethyl cation, \(^\oplus \ce{CH_2Cl}\):
Triarylmethyl Cations
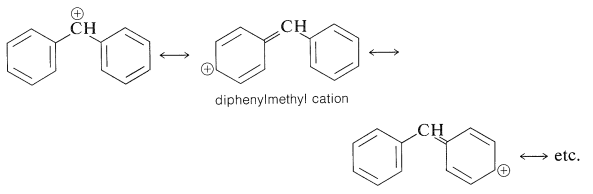
Phenylmethyl halides are similar in \(S_\text{N}1\) reactivity to 2-propenyl halides. The \(S_\text{N}1\) reactivity of phenylmethyl derivatives can be ascribed mainly to stabilization of the cation by electron delocalization. Diphenylmethyl halides, \(\ce{(C_6H_5)_2CHX}\), are still more reactive and this is reasonable because the diphenylmethyl cation has two phenyl groups over which the positive charge can be delocalized and therefore should be more stable relative to the starting halide than is the phenylmethyl cation:
Accordingly, we might expect triphenylmethyl (or trityl) halides, \(\ce{(C_6H_5)_3C-X}\), to be even more reactive. In fact, the \(\ce{C-X}\) bonds of such compounds are extremely labile. In liquid sulfur dioxide, triarylmethyl halides ionize reversibly, although the equilibria are complicated by ion-pair association:
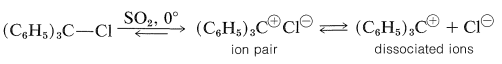
Triarylmethyl cations are among the most stable carbocations known. They are intensely colored and are formed readily when the corresponding triarylmethanols are dissolved in strong acids:
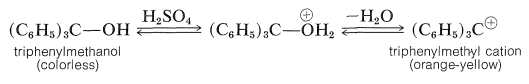
Triarylmethyl Anions
In addition to stable cations, triarylmethyl compounds form stable carbanions. Because of this the corresponding hydrocarbons are relatively acidic compared to simple alkanes. They react readily with strong bases such as sodium amide, and the resulting carbanions, like the cations, are intensely colored:

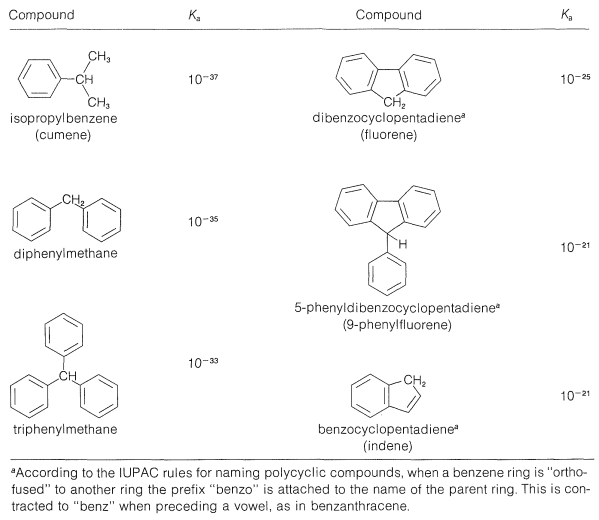
The acid strengths of arylmethanes are listed in Table 26-3. All are quite weak acids relative to water but vary over many powers of ten relative to one another. The stronger acids form the more stable carbanions, and the carbanion stability generally is determined by the effectiveness with which the negative charge can be delocalized over the substituent aryl groups.
Table 26-3: Strengths of Some Hydrocarbon Acids
Triarylmethyl Radicals
Triarylmethyl compounds also form rather stable triarylmethyl radicals, and indeed the first stable carbon free radical to be reported was the triphenylmethyl radical, \(\ce{(C_6H_5)_3C} \cdot\), prepared inadvertently by M. Gomberg in 1900. Gomberg's objective was to prepare hexaphenylethane by a Wurtz coupling reaction of triphenylmethyl chloride with metallic silver:
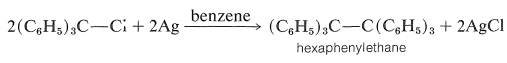
However, he found that unless air was carefully excluded from the system, the product was triphenylmethyl peroxide, \(\ce{(C_6H_5)_3COOC(C_6H_5)_3}\), rather than the expected hexaphenylethane.
Gomberg believed that the yellow solution obtained from the reaction of triphenylmethyl chloride with silver in benzene in the absence of air contained the triphenylmethyl radical. However, subsequent investigations showed that the molecular weight of the dissolved substance was closer to that for \(\ce{C_{38}H_{30}}\), hexaphenylethane. A bitter battle raged over the nature of the product and its reactions. The controversy finally was though to have been settled by the demonstration that the hydrocarbon \(\ce{C_{38}H_{30}}\) dissociates rapidly, but only slightly, to triphenylmethyl radicals at room temperature in inert solvents (\(K = 2.2 \times 10^{-4}\) at \(24^\text{o}\) in benzene). For many years thereafter, the hydrocarbon \(\ce{C_{38}H_{30}}\) was believed to be the hexaphenylethane. Now it is known that this conclusion was incorrect. The product is a dimer of triphenylmethyl, but it is formed by the addition of one radical to the 4-position of a phenyl ring of the other:

Formation of the peroxide in the presence of oxygen is explained as follows:
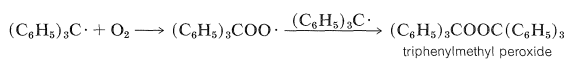
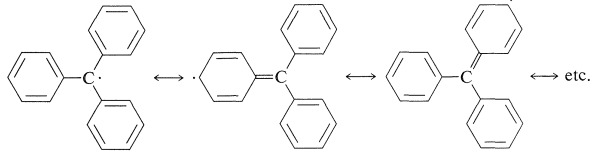
Although the foregoing reactions involving the triphenylmethyl radical seemed very unreasonable at the time they were discovered, the stability of the radical now has been established beyond question by a variety of methods such as esr spectroscopy (Section 27-9). This stability can be attributed to delocalization of the odd electron over the attached phenyl groups:
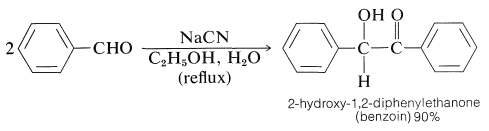
Aromatic Aldehydes. The Benzoin Condensation
Most of the reactions of aromatic aldehydes, \(\ce{ArCHO}\), are those expected of aldehydes with no \(\alpha\) hydrogens and most of these will not be reviewed here. One reaction that usually is regarded as being characteristic of aromatic aldehydes (although, in fact, it does occur with other aldehydes having no \(\alpha\) hydrogens), is known as the benzoin condensation. This reaction essentially is a dimerization fo two aldehyde molecules through the catalytic action of sodium or potassium cyanide:
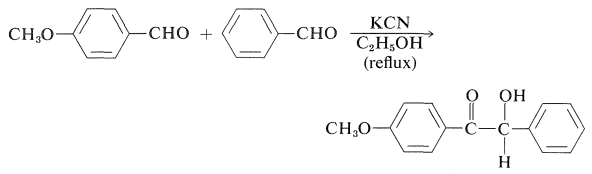
Unsymmetrical or mixed benzoins may be obtained in good yield from two different aldehydes:
As to the mechanism of benzoin formation, cyanide ion adds to the aldehyde to form \(12\). This anion is in equilibrium with \(13\), wherein the negative charge can be delocalized over the phenyl and nitrile groups. A subsequent aldol-type addition of \(13\) to the carbonyl carbon of a second aldehyde molecule gives the addition product \(14\), and loss of \(\ce{HCN}\) from \(14\) leads to the benzoin:

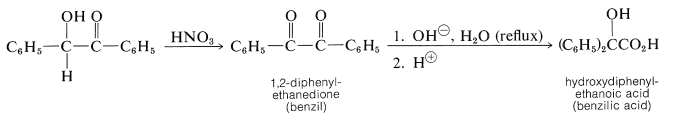
Benzoins are useful intermediates for the synthesis of other compounds because they can be oxidized to 1,2-diones and reduced in stages to various products, depending upon the reaction conditions. The 1,2-diketone known as benzil, which is obtained by nitric acid oxidation of benzoin, undergoes a base-catalyzed hydration rearrangement to form an \(\alpha\)-hydroxy acid, commonly called the benzilic acid rearrangement (see Section 17-7):
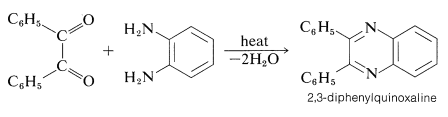
Benzils, like other 1,2-diones, react with 1,2-benzenediamines to form diazaarenes known as quinoxalines. This kind of reaction is an important general procedure for the synthesis of aromatic ring systems containing nitrogen:
Contributors and Attributions
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."