
13.3: Cycloaddition Reactions
- Page ID
- 22261

There are a variety of reactions whereby rings are formed through addition to double or triple bonds. An especially simple example is the addition of ethene to 1,3-butadiene to give cyclohexene:
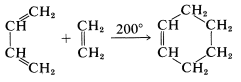
This is the prototype Diels-Alder reaction, which has proved so valuable in synthesis that it won its discoverers, O. Diels and K. Alder, the Nobel Prize in chemistry in 1950.
The Diels-Alder reaction is both a 1,4 addition or ethene to 1,3-butadiene and a 1,2 addition of butadiene to ethene. It can be called a [4 + 2] cycloaddition and as such results in the formation of a six-membered ring. Many other cycloadditions are known, such as [2 + 2], other types of [4 + 2], and [2 + 2 + 2], which give different size of rings. Some specific examples follow:

The synthetic importance of these reactions is very great and, because many of them often involve dienes, we will discuss their general characteristics in this chapter. The most valuable cycloaddition reaction almost certainly is the [4 + 2], or Diels-Alder, reaction and will be discussed in detail.
[4 + 2] Cycloadditions
There is one very important point you should remember about the Diels-Alder reaction: The reaction usually occurs well only when the [2] component is substituted with electron-attracting groups and the [4] component is substituted with electron-donating groups, or the reverse. The most common arrangement is to have the alkene (usually referred to as the dienophile) substituted with electron-attracting groups such as \(\ce{-CO_2H}\), \(\ce{-COR}\), or \(\ce{-C \equiv N}\). For example,
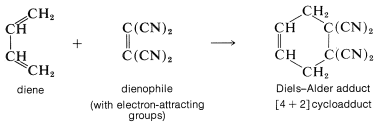
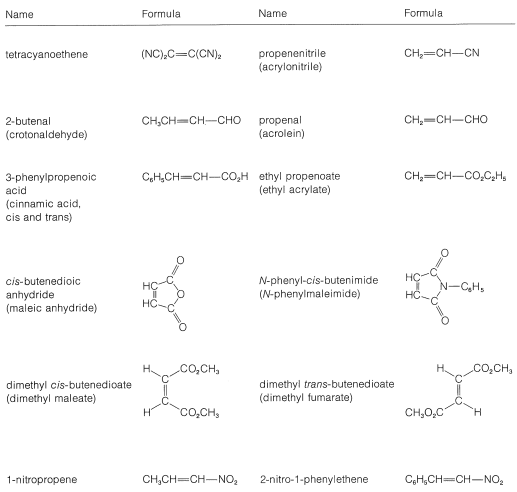
A list of the more reactive dienophiles carrying electron-attracting groups is given in Table 13-1. Ethene and other simple alkenes generally are poor dienophiles and react with 1,3-butadiene only under rather extreme conditions and in low yield.
However, when the diene is substituted with several electron-attracting groups such as chlorine or bromine, electron-donating groups on the dienophile facilitate the reaction. Many substances, such as 2-methylpropene, that act as dienophiles with hexachlorocyclopentadiene simply will not undergo [4 + 2] addition with cyclopentadiene itself:
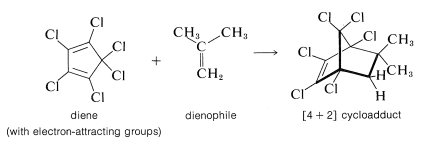
Table 13-1: Reactive Dienophiles with 1,3-Butadiene and Similar Dienes
The Diels-Alder reaction is highly stereospecific. The diene reacts in an unfavorable conformation in which its double bonds lie in a plane on the same side (cis) of the single bond connecting them. This s-cis (or cisoid) conformation is required to give a stable product with a cis double bond. Addition of ethene to the alternate and more stable (transoid) conformation would give an impossibly strained trans-cyclohexene ring. Possible transition states for reaction in each conformation follow, and it will be seen that enormous molecular distortion would have to take place to allow addition of ethene to the transoid conformation:
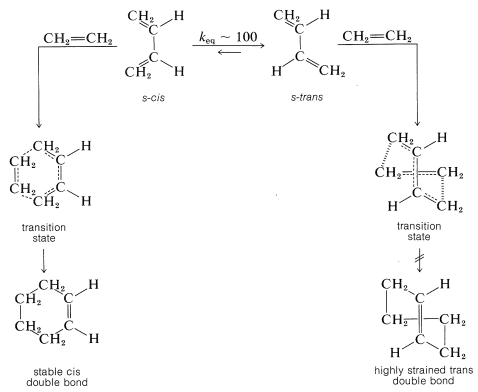
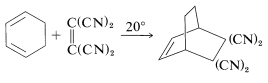
Cyclic dienes usually react more readily than open-chain dienes, probably because they have their double bonds fixed in the proper conformation for [4 + 2] cycloaddition, consequently the price in energy of achieving the s-cis configuration already has been paid:
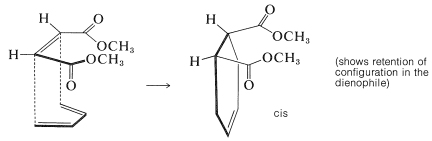
Further evidence of stereospecificity in [4 + 2] additions is that the configurations of the diene and the dienophile are retained in the adduct. This means that the reactants (or addends) come together to give suprafacial addition. Two examples follow, which are drawn to emphasize how suprafacial addition occurs. In the first example, dimethyl cis-butadioate adds to 1,3-butadiene to give a cis-substituted cyclohexene:
In the second example, suprafacial approach of a dienophile to the 2,5 carbons of trans,trans-2,4-hexadiene is seen to lead to a product with two methyl groups on the same side of the cyclohexene ring:
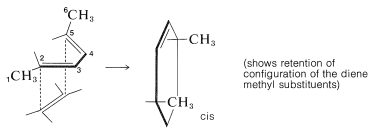
(The use of models will help you visualize these reactions and their stereochemistry.)
There is a further feature of the Diels-Alder reaction that concerns the stereochemical orientation of the addends. In the addition of cis-butanedioic anhydride (maleic anhydride) to cyclopentadiene there are two possible ways that the diene and the dienophile could come together to produce different products. These are shown in Equations 13-3 and 13-4:
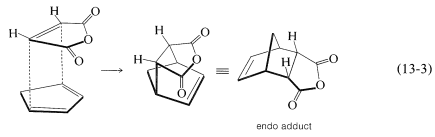
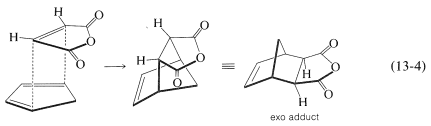
In practice, the adduct with the endo\(^2\) configuration usually is the major product. As a general rule, Diels-Alder additions tend to proceed to favor that orientation that corresponds to having the diene double bonds and the unsaturated substituents of the dienophile closest to one another. This means that addition by Equation 13-3 is more favorable than by Equation 13-4, but the degree of endo-exo stereospecificity is not as high as the degree of stereospecificity of suprafacial addition to the diene and dienophile.
There are exceptions to favored endo stereochemistry of Diels-Alder additions. Some of these exceptions arise because the addition reaction is reversible, dissociation being particularly important at high temperature. The exo configuration is generally more stable than the endo and, given time to reach equilibrium (cf. Section 10-4A), the exo isomer may be the major adduct. Thus endo stereospecificity can be expected only when the additions are subject to kinetic control.
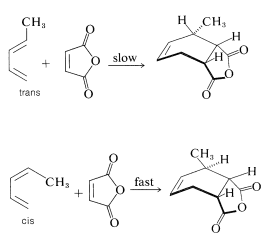
The reactivities of dienes in the Diels-Alder reaction depend on the number and kind of substituents they possess. The larger the substituents are, or the more of them, at the ends of the conjugated system, the slower the reaction is likely to be. There also is a marked difference in reactivity with diene configuration. Thus trans-1,3-pentadiene is substantially less reactive toward a given dienophile (such as maleic anhydride) than is cis-1,3-pentadiene. In fact, a mixture of the cis and trans isomers can be separated by taking advantage of the difference in their reactivities on cycloaddition:
Mechanism of the Diels-Alder Reaction
There is little evidence to support simple radical or polar mechanisms (such as we have discussed previously) for the Diels-Alder reaction. As the result of many studies the reaction seems best formulated as a process in which the bonds between the diene and the dienophile are formed essentially simultaneously:
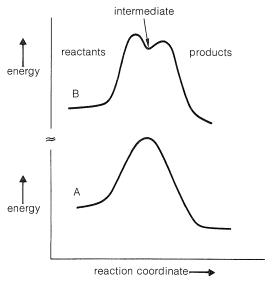
We already have discussed a few addition reactions that appear to occur in a concerted manner. These include the addition of diimide, ozone, and boron hydrides to alkenes (Sections 11-5, 11-7A, and 11-6B). Concerted reactions that have cyclic transition states often are called pericyclic reactions. Other examples will be considered in later chapters.
A [4 + 2] Cycloaddition
We indicated previously that sulfur dioxide \(\left( \ce{SO_2} \right)\) and 1,3-butadiene form a [4 + 1] cycloaddition product:
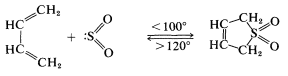
This reaction is more readily reversible than most Diels-Alder reactions, and the product largely dissociates to the starting materials on heating to \(120^\text{o}\). The cycloadduct is an unsaturated cyclic sulfone, which can be hydrogenated to give the saturated cyclic sulfone known as "sulfolane":
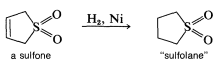
This compound is used extensively in the petrochemical industry as a selective solvent.
The reversibility of the diene-\(\ce{SO_2}\) cycloaddition makes it useful in the purification of reactive dienes. 2-Methyl-1,3-butadiene (isoprene) is purified commercially in this manner prior to being polymerized to rubber (Section 13-4):
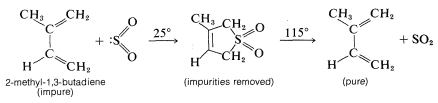
Neither 1,3-cyclopentadiene nor 1,3-cyclohexadiene react with sulfur dioxide, probably because the adducts would be too highly strained:
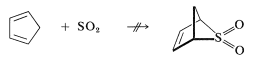
Some [2 + 2] Cycloadditions
Many naturally occurring organic compounds contain six-membered carbon rings, but there are relatively few with four-membered carbon rings. After encountering the considerable ease with which six-membered rings are formed by [4 + 2] cycloaddition, we might expect that the simpler [2 + 2] cycloadditions to give four-membered rings also should go well, provided that strain is not too severe in the products. In fact, the dimerization of ethene is thermodynamically favorable:
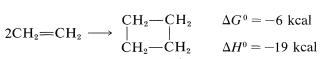
Nonetheless, this and many other [2 + 2] cycloaddition reactions do not occur on simple heating.
However, there are a few exceptions. One is the dimerization of tetrafluoroethene, which perhaps is not surprising, considering the favorable thermodynamic parameters:
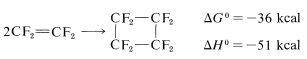
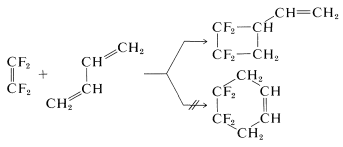
What is surprising is that addition of \(\ce{CF_2=CF_2}\) to 1,3-butadiene gives a cyclobutane and not a cyclohexane, although the [2 + 2] product probably is about \(25 \: \text{kcal mol}^{-1}\) less stable than the [4 + 2] product:
Such [2 + 2] thermal additions generally are limited to polyhaloethenes and a few substances with cumulated double bonds, such as 1,2-propadiene \(\left( \ce{CH_2=C=CH_2} \right)\) and ketenes \(\left( \ce{R_2C=C=O} \right)\). Some examples follow:
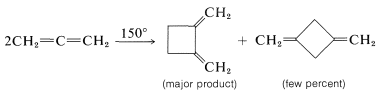
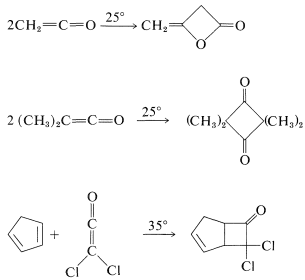
Many [2 + 2] cycloadditions that do not occur by simply heating the possible reactants can be achieved by irradiation with ultraviolet light. The following example, [2 + 2] addition of 2-cyclopentenone to cyclopentene, occurs photochemically but not thermally:
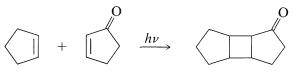
In all such photochemical cycloadditions the energy required to achieve a cycloaddition transition state, which can amount to \(100 \: \text{kcal mol}^{-1}\) or more, is acquired by absorption of light.
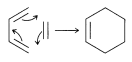
Thermodynamically unfavorable cycloaddition products can be formed photochemically. A striking example is the photochemical conversion of norbornadiene to quadricyclene. The reverse of this reaction can occur with almost explosive violence in the presence of appropriate metal catalysts or on simple heating:

Why do some [2 + 2] cycloadditions occur thermally and others photochemically? What is special about fluoroalkenes and cumulated dienes? The answers are complex, but it appears that most thermal [2 + 2] cycloadditions, unlike the Diels-Alder [4 + 2] cycloadditions, go by stepwise routes (see Section 21-11). Why the two types of thermal cycloaddition have different mechanisms will be discussed in Sections 21-10A and B.
\(^2\)In general, the designation endo or exo refers to configuration in bridged or polycyclic ring systems such as those shown in Equations 13-3 and 13-4. With reference to the bridge atoms, a substituent is exo if it is on the same side as the bridge, or endo if it is on the opposite side. Further examples are
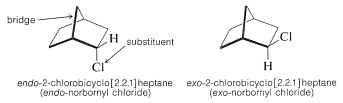
In drawing endo and exo isomers, it is best to represent the actual spatial relationships of the atoms as closely as possible. The cyclohexane ring is shown here in the boat form (Section 12-3A) because it is held in this configuration by the methylene group that bridges the 1,4 positions. If you do not see this, we strongly advise that you construct models.
Contributors and Attributions
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."
- Layne A. Morsch (University of Illinois Springfield)