
1.26: Alkenes and Alkyne Structure
- Page ID
- 81792

Alkene Double Bond Structure
Today we'll begin by looking at the structural characteristics of alkenes, the products of elimination reactions. Then we'll return to the topic of elimination reactions and examine their reaction mechanisms in more detail.
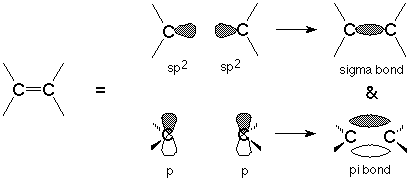
The functional group of an alkene is the carbon-carbon double bond. In common with the double bond we studied at the beginining of the course (the carbon-oxygen double bond in the carbonyl group) this double bond consists of a sigma bond, viewed as "end to end" overlap between sp2 hybridized orbitals on the carbon atoms, and a pi bond, viewed as "side to side" overlap between the p orbitals on the same carbon atoms.
Stereoisomerism
The presence of the pi bond in the alkene functional group has two important consequences. First, the reactions of alkenes are essentially the reactions of this pi bond. We'll look at these reactions in some detail next time. Second, the pi bond means that rotation of one end of the double bond relative to the other requires so much energy that it does not happen at ordinary temperatures. This is because such rotation would destroy the "side to side" overlap of the p orbitals which make up the pi bond and would effectively break the pi bond. Breaking bonds requires energy input.
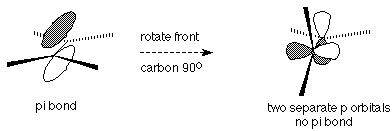
The result of this can be seen in the fact that there are two different substances which have the same "connectivity" structure and are both called 2-butene. (Alkene naming is treated in Section 5.2 of Brown). A sample of one of these compounds does not become the other, since to do so would require breaking the pi bond and there isn't enough energy available to do that. Since the difference is one of spatial arrangement, this is a type of stereoisomerism.

The absence of rotation about the double bond explains why there are two different 2-butene molecules. These differences must be reflected in the names given to the compounds. If there are two identical groups, one on each carbon involved in the double bond, then we can use the terms cis (which means that the two identical groups -- hydrogens or methyls in this case -- are on the same side of the double bond) or trans (means that the two identical groups are on opposite sides of the double bond).
This system breaks down when there are not identical groups on the two carbon atoms. In this type of situation we use a system which is derived from the same ranking rules we used in assigning R and S configurations to stereogenic carbon atoms. To use this system we look at each of the two doubly bonded carbons independently. For one carbon we examine the two groups or atoms which are connected to it by single bonds. We use the ranking rules to decide which of these groups or atoms has the higher ranking. Then weapply the same process to the second carbon atom. If the higher ranked group on one carbon is on the same side of the double bond as the higher ranked group on the other carbon, then the designation is Z (from the German word "Zusammen"). If the higher ranked group on one carbon is on the other side from the higher ranked group on the other carbon, the designation is E (from "Entgegen"). Here's an example:
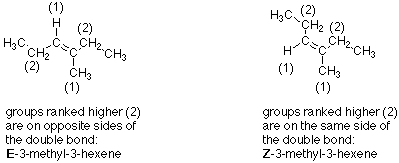
Like all naming situations, you learn by doing problems, so do the naming problems listed in the internet syllabus and bring up points that need clarification. See Section 5.2C in Brown for further explanation.
Elimination Reactions
Now let's turn our attention to making this double bond. The reactions which do this are called elimination reactions, because two atoms or groups are "eliminated" from an alkyl halide or alcohol so that the double bond can be formed. A formal example is:

Notice that the two atoms eliminated were attached to adjacent carbon atoms. This must be so if we are to make an new pi bond between those atoms. If the atom bearing the bromine is designated the alpha carbon atom, then the one next to it is a beta carbon atom. These eliminations are often called beta eliminations.
In many cases there are more than one beta carbon atom. This can lead to situations where more than one beta-elimination product is possible. Here's an example:
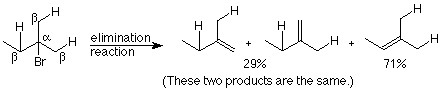
Notice that the major product is the one which has the most substituents (non-hydrogen atoms, in this case, methyl groups) attached to the doubly bonded carbons. This is generally the case, and it is called Zaitsev's rule after Alexander Zaitsev the 19th century Russian chemist who first proposed the general statement. In elimination reactions the major product is the one in which the maximum number of substituents is attached to the doubly bonded carbons. (Notice that the carbon skeleton is not changed in this reaction.) Zaitsev's rule enables us to predict which of two or more possible beta-elimination products can be expected to predominate.
Often we can choose an alkyl halide such that only one beta-elimination product is possible. For example, if we wanted to make the "minor" product from the example above, we might choose the following approach:
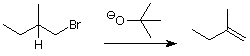
Here we've chosen an alkyl halide which has only one beta hydrogen, so it can give only one alkene in a beta-elimination reaction. We've also chosen a base which is bulky to slow down the competing substitution reaction.
The E2 Mechanism - Dehydrohalogenation
The most important elimination reaction mechanisms are closely related to substitution mechanisms. The first one we'll study is called the E2 mechanism, E for elimination and 2 for second order. In studying the SN2 mechanism we learned that second order meant that the concentrations of both the alkyl halide and the nucleophile were important in determining the rate of the reaction.
rate = k[RX]{Nu]
We interpreted this to mean that the alkyl halide and the nucleophile must collide in order to form the transition state, which smoothly completes the reaction and becomes the product. The same interpretation occurs here. The important difference is that the reactant we considered to be a nucleophile attacking carbon in the SN2 reaction is now acting as a base and attacking a beta hydrogen.
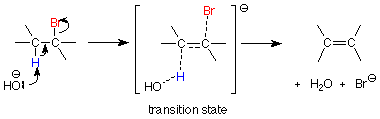
If we follow the curved arrow notation we see that formation of the O-H bond releases the C-H bonding electrons to begin to form the new C-C pi bond. The C-Br bond breaks at the same time to provide room for the new C-C pi bond to develop. All of these bonding changes occur in one step. The transition state (top of the energy hill) has three partial bonds in it. Since it includes both the alkyl halide and the base, it is consistent with the need for them to collide in order for the reaction to occur and with the second order kinetic behavior.
Since the transition state structure has a partial broken C-Br bond just like the partially broken C-Br bond in the SN2 transition state, we expect that changing from one halide to another will produce the same change in rate that we saw for the SN2 mechanism:
RI > RBr > RCl > RF
This is what is observed.
Since the base plays such an essential role in this mechanism, it is very likely that the elimination reactions we considered to be unwanted complications when we were studying SN2 reactions were in fact E2 reactions. Recall that we learned that these eliminations are most prevalent when we have strong base (essential for an E2 mechanism) and a tertiary alkyl halide (which would make the competing SN2 mechanism very slow). The combination would be good for a fast E2 elimination and a slow SN2 substitution and would predict lots of elimination product.
The E2 mechanism is very closely related to the SN2 mechanism. In much the same way, there is an E1 mechanism which involves the same first step and the same carbocation intermediate as the SN1 mechanism. Here's the pattern:
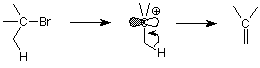
Notice that the first step is identical to the first step in the SN1: dissociation of a halide ion to form a carbocation. The E1 mechanism continues by loss of a proton and the formation of a new pi bond. The first step determines how fast the reaction goes. The relative proportions of substistution and elimination product are determined by the relative rates of nucleophilic attack on the carbocation carbon (substitution by SN1) or loss of the proton (elimination by E1).
Since the rate of reactions proceeding by this mechanism is determined by the rate at which the carbocation is formed, the effect of changing alkyl group structure is the same as it was for the SN1 mechanism. (Notice that a methyl halide only has one carbon and cannot make a double bond.)
tertiary > secondary > primary
Notice that the rate of this reaction does not depend upon the base concentration. The base is not involved until after the carbocation is formed in the rate determining step. This means that the E1 mechanism is likely to be used where no strong base is present.
The E1 Mechanism - Dehydration
Alcohols can also be used to make alkenes by elimination reactions. We know that the OH- group is a poor leaving group (it's a strong base and strong bases make poor leaving groups). To permit an alcohol to react by breaking the C-O bond, the OH group must first be changed into a better leaving group. This can be done using an acid to place a proton on the OH oxygen. This makes an H2O of the OH, and since water is a much weaker base than OH-, it is a much better leaving group.
Once the alcohol has been protonated, what happens next depends upon the alkyl group structure and what else is in the reaction mixture. If there are bromide or chloride ions present (perhaps because the acid used was HBr or HCl) then those bromide or chloride ions can serve as good nucleophiles and complete a substitution reaction. If we have used only a small, catalytic, amount of an acid such as sulfuric acid, then there is only a small amount of bisulfate ion present to act as a nucleophile. The reaction is more likely to result in elimination. One important factor in this outcome is heating the reaction. Alkenes have lower boiling points than alcohols, so once an alkene is produced, it boils out of the reaction mixture and is collected by distillation. Removing the alkene as it is formed protects it from other possible reactions.
If the alcohol is primary an elimination uses the E2 pathway, primarily since forming primary carbocations is so slow. Ethylene can be made this way. Notice the close resemblance to the E2 mechanism for alky halides, with the major difference that no strong base is involved here since a strong base cannot exist in an acid solution.
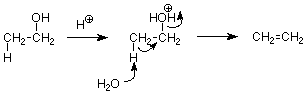
Secondary and tertiary alcohols are more likely to use the E1 pathway. Here's a typical mechanism:
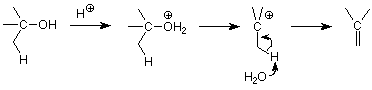
Like alkyl halide dehydrohalogenation (elimination), dehydration of alcohols follows Zaitsev's rule -- the more highly substituted alkene is the major product.