
14.7: Reaction Directions (Thermodynamic Explanation)
- Page ID
- 41619
- To know the relationship between free energy and the equilibrium constant.
We have identified three criteria for whether a given reaction will occur spontaneously:
- \(ΔS_{univ} > 0\) (2nd law of thermodynamics),
- \(ΔG_{sys} < 0\) (Gibbs Energy version), and
- the relative magnitude of the reaction quotient \(Q\) versus the equilibrium constant \(K\).
Recall that if \(Q < K\), then the reaction proceeds spontaneously to the right as written, resulting in the net conversion of reactants to products. Conversely, if \(Q > K\), then the reaction proceeds spontaneously to the left as written, resulting in the net conversion of products to reactants. If \(Q = K\), then the system is at equilibrium, and no net reaction occurs. Table \(\PageIndex{1}\) summarizes these criteria and their relative values for spontaneous, nonspontaneous, and equilibrium processes.
| Spontaneous | Equilibrium | Nonspontaneous* |
|---|---|---|
| *Spontaneous in the reverse direction. | ||
| ΔSuniv > 0 | ΔSuniv = 0 | ΔSuniv < 0 |
| ΔGsys < 0 | ΔGsys = 0 | ΔGsys > 0 |
| Q < K | Q = K | Q > K |
Because all three criteria assess the same thing—the spontaneity of the process—it would be most surprising indeed if they were not related. In this section, we explore the relationship between the standard free energy of reaction (\(ΔG°\)) and the equilibrium constant (\(K\)).
Free Energy and the Equilibrium Constant
Because ΔH° and ΔS° determine the magnitude of ΔG° and because K is a measure of the ratio of the concentrations of products to the concentrations of reactants, we should be able to express K in terms of ΔG° and vice versa. "Free Energy", ΔG is equal to the maximum amount of work a system can perform on its surroundings while undergoing a spontaneous change. For a reversible process that does not involve external work, we can express the change in free energy in terms of volume, pressure, entropy, and temperature, thereby eliminating \(ΔH\) from the equation for \(ΔG\). The general relationship can be shown as follow (derivation not shown):
\[ \Delta G = V \Delta P − S \Delta T \label{18.29}\]
If a reaction is carried out at constant temperature (\(ΔT = 0\)), then Equation \(\ref{18.29}\) simplifies to
\[\Delta{G} = V\Delta{P} \label{18.30}\]
Under normal conditions, the pressure dependence of free energy is not important for solids and liquids because of their small molar volumes. For reactions that involve gases, however, the effect of pressure on free energy is very important.
Assuming ideal gas behavior, we can replace the \(V\) in Equation \(\ref{18.30}\) by nRT/P (where n is the number of moles of gas and R is the ideal gas constant) and express \(\Delta{G}\) in terms of the initial and final pressures (\(P_i\) and \(P_f\), respectively):
\[\begin{align} \Delta G &=\left(\dfrac{nRT}{P}\right)\Delta P \\[4pt] &=nRT\dfrac{\Delta P}{P}=nRT\ln\left(\dfrac{P_\textrm f}{P_\textrm i}\right) \label{18.31} \end{align} \]
If the initial state is the standard state with Pi = 1 atm, then the change in free energy of a substance when going from the standard state to any other state with a pressure P can be written as follows:
\[G − G^° = nRT\ln{P}\]
This can be rearranged as follows:
\[G = G^° + nRT\ln {P} \label{18.32}\]
As you will soon discover, Equation \(\ref{18.32}\) allows us to relate \(ΔG^o\) and \(K_p\). Any relationship that is true for \(K_p\) must also be true for \(K\) because \(K_p\) and \(K\) are simply different ways of expressing the equilibrium constant using different units.
Let’s consider the following hypothetical reaction, in which all the reactants and the products are ideal gases and the lowercase letters correspond to the stoichiometric coefficients for the various species:
\[aA+bB \rightleftharpoons cC+dD \label{18.33}\]
Because the free-energy change for a reaction is the difference between the sum of the free energies of the products and the reactants, we can write the following expression for \(ΔG\):
\[\begin{align} \Delta{G}&=\sum_m G_{products}−\sum_n G_{reactants} \\[4pt] &=(cG_C+dG_D)−(aG_A+bG_B) \label{18.34} \end{align} \]
Substituting Equation \(\ref{18.32}\) for each term into Equation \(\ref{18.34}\),
\[ΔG=[(cG^o_C+cRT \ln P_C)+(dG^o_D+dRT\ln P_D)]−[(aG^o_A+aRT\ln P_A)+(bG^o_B+bRT\ln P_B)]\]
Combining terms gives the following relationship between \(ΔG\) and the reaction quotient \(Q\):
\[\begin{align} \Delta G &=\Delta G^\circ+RT \ln\left(\dfrac{P^c_\textrm CP^d_\textrm D}{P^a_\textrm AP^b_\textrm B}\right) \\[4pt] &=\Delta G^\circ+RT\ln Q \label{18.35} \end{align}\]
where \(ΔG°\) indicates that all reactants and products are in their standard states. For gases at equilibrium (\(Q = K_p\),), and as you’ve learned in this chapter, \(ΔG = 0\) for a system at equilibrium. Therefore, we can describe the relationship between \(ΔG^o\) and \(K_p\) for gases as follows:
\[ \begin{align} 0 &= ΔG° + RT\ln K_p \label{18.36a} \\[4pt] ΔG° &= −RT\ln K_p \label{18.36b} \end{align} \]
If the products and reactants are in their standard states and ΔG° < 0, then Kp > 1, and products are favored over reactants. Conversely, if ΔG° > 0, then Kp < 1, and reactants are favored over products. If ΔG° = 0, then \(K_p = 1\), and neither reactants nor products are favored: the system is at equilibrium.
For a spontaneous process under standard conditions, \(K_{eq}\) and \(K_p\) are greater than 1.
To further illustrate the relation between these two essential thermodynamic concepts, consider the observation that reactions spontaneously proceed in a direction that ultimately establishes equilibrium. As may be shown by plotting the free energy change versus the extent of the reaction (for example, as reflected in the value of Q), equilibrium is established when the system’s free energy is minimized (Figure \(\PageIndex{3}\)). If a system is present with reactants and products present in nonequilibrium amounts (Q ≠ K), the reaction will proceed spontaneously in the direction necessary to establish equilibrium.
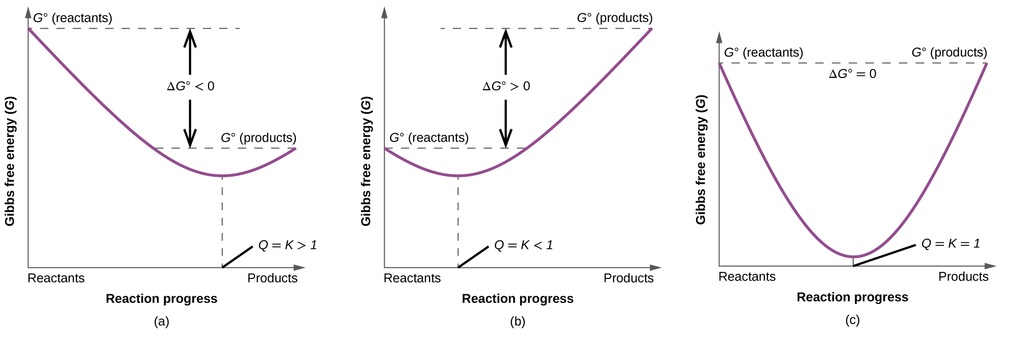
\(ΔG^o\) is −32.7 kJ/mol of N2 for the reaction
\[\ce{N2(g) + 3H2(g) \rightleftharpoons 2NH3(g)}\nonumber\]
This calculation was for the reaction under standard conditions—that is, with all gases present at a partial pressure of 1 atm and a temperature of 25°C. Calculate \(ΔG\) for the same reaction under the following nonstandard conditions:
- \(P_{\textrm N_2}\) = 2.00 atm,
- \(P_{\textrm H_2}\) = 7.00 atm,
- \(P_{\textrm{NH}_3}\) = 0.021 atm,
- and T = 100°C.
Does the reaction favor products or reactants?
Given: balanced chemical equation, partial pressure of each species, temperature, and ΔG°
Asked for: whether products or reactants are favored
Strategy:
- Using the values given and Equation \(\ref{18.35}\), calculate \(Q\).
- Substitute the values of ΔG° and Q into Equation \(\ref{18.35}\) to obtain \(ΔG\) for the reaction under nonstandard conditions.
Solution:
A The relationship between ΔG° and ΔG under nonstandard conditions is given in Equation \(\ref{18.35}\). Substituting the partial pressures given, we can calculate \(Q\):
\[Q=\dfrac{P^2_{\textrm{NH}_3}}{P_{\textrm N_2}P^3_{\textrm H_2}}=\dfrac{(0.021)^2}{(2.00)(7.00)^3}=6.4\times10^{-7}\nonumber\]
B Substituting the values of ΔG° and Q into Equation \(\ref{18.35}\),
\[ \begin{align*} \Delta G &=\Delta G^\circ+RT\ln Q \\[4pt] &=-32.7\textrm{ kJ}+\left[(\textrm{8.314 J/K})(\textrm{373 K})\left(\dfrac{\textrm{1 kJ}}{\textrm{1000 J}}\right)\ln(6.4\times10^{-7})\right] \\[4pt] &=-32.7\textrm{ kJ}+(-44\textrm{ kJ}) \\[4pt] &=-77\textrm{ kJ/mol of N}_2 \end{align*}\]
Because ΔG < 0 and Q < 1.0, the reaction is spontaneous to the right as written, so products are favored over reactants.
Calculate \(ΔG\) for the reaction of nitric oxide with oxygen to give nitrogen dioxide under these conditions: T = 50°C, PNO = 0.0100 atm, \(P_{\mathrm{O_2}}\) = 0.200 atm, and \(P_{\mathrm{NO_2}}\) = 1.00 × 10−4 atm. The value of ΔG° for this reaction is −72.5 kJ/mol of O2. Are products or reactants favored?
- Answer
-
−92.9 kJ/mol of O2; the reaction is spontaneous to the right as written, so products are favored.
Calculate Kp for the reaction of H2 with N2 to give NH3 at 25°C. \(ΔG^o\) for this reaction is −32.7 kJ/mol of N2.
Given: balanced chemical equation from Example \(\PageIndex{2}\), ΔG°, and temperature
Asked for: Kp
Strategy:
Substitute values for ΔG° and T (in kelvin) into Equation \(\ref{18.36b}\) to calculate \(K_p\), the equilibrium constant for the formation of ammonia.
Solution
In Example 10, we used tabulated values of ΔG∘f to calculate ΔG° for this reaction (−32.7 kJ/mol of N2). For equilibrium conditions, rearranging Equation \(\ref{18.36b}\),
\[\begin{align*} \Delta G^\circ &=-RT\ln K_\textrm p \\[4pt] \dfrac{-\Delta G^\circ}{RT} &=\ln K_\textrm p \end{align*}\]
Inserting the value of ΔG° and the temperature (25°C = 298 K) into this equation,
\[\begin{align*}\ln K_\textrm p &=-\dfrac{(-\textrm{32.7 kJ})(\textrm{1000 J/kJ})}{(\textrm{8.314 J/K})(\textrm{298 K})}=13.2 \\[4pt] K_\textrm p &=5.4\times10^5\end{align*}\]
Thus the equilibrium constant for the formation of ammonia at room temperature is favorable. However, the rate at which the reaction occurs at room temperature is too slow to be useful.
Calculate Kp for the reaction of NO with O2 to give NO2 at 25°C. ΔG° for this reaction is −70.5 kJ/mol of O2.
- Answer
-
2.2 × 1012
Although \(K_p\) is defined in terms of the partial pressures of the reactants and the products, the equilibrium constant \(K\) is defined in terms of the concentrations of the reactants and the products. We described the relationship between the numerical magnitude of \(K_p\) and \(K\) previously and showed that they are related:
\[K_p = K(RT)^{Δn} \label{18.37}\]
where \(Δn\) is the number of moles of gaseous product minus the number of moles of gaseous reactant. For reactions that involve only solutions, liquids, and solids, \(Δn = 0\), so \(K_p = K\). For all reactions that do not involve a change in the number of moles of gas present, the relationship in Equation \(\ref{18.36b}\) can be written in a more general form:
\[ΔG° = −RT \ln K \label{18.38}\]
Only when a reaction results in a net production or consumption of gases is it necessary to correct Equation \(\ref{18.38}\) for the difference between \(K_p\) and \(K\). Although we typically use concentrations or pressures in our equilibrium calculations, recall that equilibrium constants are generally expressed as unitless numbers because of the use of activities or fugacities in precise thermodynamic work. Systems that contain gases at high pressures or concentrated solutions that deviate substantially from ideal behavior require the use of fugacities or activities, respectively.
Combining Equations \(\ref{18.38}\) with \(ΔG^o = ΔH^o − TΔS^o\) provides insight into how the components of ΔG° influence the magnitude of the equilibrium constant:
\[ΔG° = ΔH° − TΔS° = −RT \ln K \label{18.39}\]
Notice that \(K\) becomes larger as ΔS° becomes more positive, indicating that the magnitude of the equilibrium constant is directly influenced by the tendency of a system to move toward maximum disorder. Moreover, K increases as ΔH° decreases. Thus the magnitude of the equilibrium constant is also directly influenced by the tendency of a system to seek the lowest energy state possible.
The magnitude of the equilibrium constant is directly influenced by the tendency of a system to move toward maximum disorder and seek the lowest energy state possible.
Temperature Dependence of the Equilibrium Constant
The fact that ΔG° and K are related provides us with another explanation of why equilibrium constants are temperature dependent. This relationship is shown explicitly in Equation \(\ref{18.39}\), which can be rearranged as follows:
\[\ln K=-\dfrac{\Delta H^\circ}{RT}+\dfrac{\Delta S^\circ}{R} \label{18.40}\]
Assuming ΔH° and ΔS° are temperature independent, for an exothermic reaction (ΔH° < 0), the magnitude of K decreases with increasing temperature, whereas for an endothermic reaction (ΔH° > 0), the magnitude of K increases with increasing temperature. The quantitative relationship expressed in Equation \(\ref{18.40}\) agrees with the qualitative predictions made by applying Le Chatelier’s principle. Because heat is produced in an exothermic reaction, adding heat (by increasing the temperature) will shift the equilibrium to the left, favoring the reactants and decreasing the magnitude of K. Conversely, because heat is consumed in an endothermic reaction, adding heat will shift the equilibrium to the right, favoring the products and increasing the magnitude of K. Equation \(\ref{18.40}\) also shows that the magnitude of ΔH° dictates how rapidly K changes as a function of temperature. In contrast, the magnitude and sign of ΔS° affect the magnitude of K but not its temperature dependence.
If we know the value of K at a given temperature and the value of ΔH° for a reaction, we can estimate the value of K at any other temperature, even in the absence of information on ΔS°. Suppose, for example, that K1 and K2 are the equilibrium constants for a reaction at temperatures T1 and T2, respectively. Applying Equation \(\ref{18.40}\) gives the following relationship at each temperature:
\[\begin{align}\ln K_1&=\dfrac{-\Delta H^\circ}{RT_1}+\dfrac{\Delta S^\circ}{R} \\[4pt] \ln K_2 &=\dfrac{-\Delta H^\circ}{RT_2}+\dfrac{\Delta S^\circ}{R}\end{align}\]
Subtracting \(\ln K_1\) from \(\ln K_2\),
\[\ln K_2-\ln K_1=\ln\dfrac{K_2}{K_1}=\dfrac{\Delta H^\circ}{R}\left(\dfrac{1}{T_1}-\dfrac{1}{T_2}\right) \label{18.41}\]
Thus calculating ΔH° from tabulated enthalpies of formation and measuring the equilibrium constant at one temperature (K1) allow us to calculate the value of the equilibrium constant at any other temperature (K2), assuming that ΔH° and ΔS° are independent of temperature. Equation \(\ref{18.41}\) is often referred to as the van 't Hoff equation after Dutch chemist Jacobus Henricus van 't Hoff in 1884 in his book Études de Dynamique chimique (Studies in dynamic chemistry).
The equilibrium constant for the formation of NH3 from H2 and N2 at 25°C was calculated to be Kp = 5.4 × 105 in Example \(\PageIndex{2}\). What is Kp at 500°C? (Use the data from Example \(\PageIndex{1}\))
Given: balanced chemical equation, ΔH°, initial and final T, and Kp at 25°C
Asked for: Kp at 500°C
Strategy:
Convert the initial and final temperatures to kelvin. Then substitute appropriate values into Equation \(\ref{18.41}\) to obtain \(K_2\), the equilibrium constant at the final temperature.
Solution:
The value of ΔH° for the reaction obtained using Hess’s law is −91.8 kJ/mol of N2. If we set \(T_1\) = 25°C = 298 K and \(T_2\) = 500°C = 773 K, then from Equation \(\ref{18.41}\) we obtain the following:
\[\begin{align*}\ln\dfrac{K_2}{K_1}&=\dfrac{\Delta H^\circ}{R}\left(\dfrac{1}{T_1}-\dfrac{1}{T_2}\right) \\[4pt] &=\dfrac{(-\textrm{91.8 kJ})(\textrm{1000 J/kJ})}{\textrm{8.314 J/K}}\left(\dfrac{1}{\textrm{298 K}}-\dfrac{1}{\textrm{773 K}}\right)=-22.8 \\[4pt] \dfrac{K_2}{K_1}&=1.3\times10^{-10} \\[4pt] K_2&=(5.4\times10^5)(1.3\times10^{-10})=7.0\times10^{-5}\end{align*}\]
Thus at 500°C, the equilibrium strongly favors the reactants over the products.
The equilibrium constant for the reaction of \(\ce{NO}\) with \(\ce{O2}\) to give \(\ce{NO2}\) at 25°C is \(K_p = 2.2 \times 10{12}\). Use the \(ΔH^o_f\) values in the exercise in Example \(\PageIndex{4}\) to calculate \(K_p\) for this reaction at 1000°C.
- Answer
-
5.6 × 10−4
Summary
For a reversible process that does not involve external work, we can express the change in free energy in terms of volume, pressure, entropy, and temperature. If we assume ideal gas behavior, the ideal gas law allows us to express \(ΔG\) in terms of the partial pressures of the reactants and products, which gives us a relationship between ΔG and Kp, the equilibrium constant of a reaction involving gases, or K, the equilibrium constant expressed in terms of concentrations. If ΔG° < 0, then K or Kp > 1, and products are favored over reactants. If ΔG° > 0, then K or Kp < 1, and reactants are favored over products. If ΔG° = 0, then K or Kp = 1, and the system is at equilibrium. We can use the measured equilibrium constant K at one temperature and ΔH° to estimate the equilibrium constant for a reaction at any other temperature.
Contributors and Attributions
- Anonymous