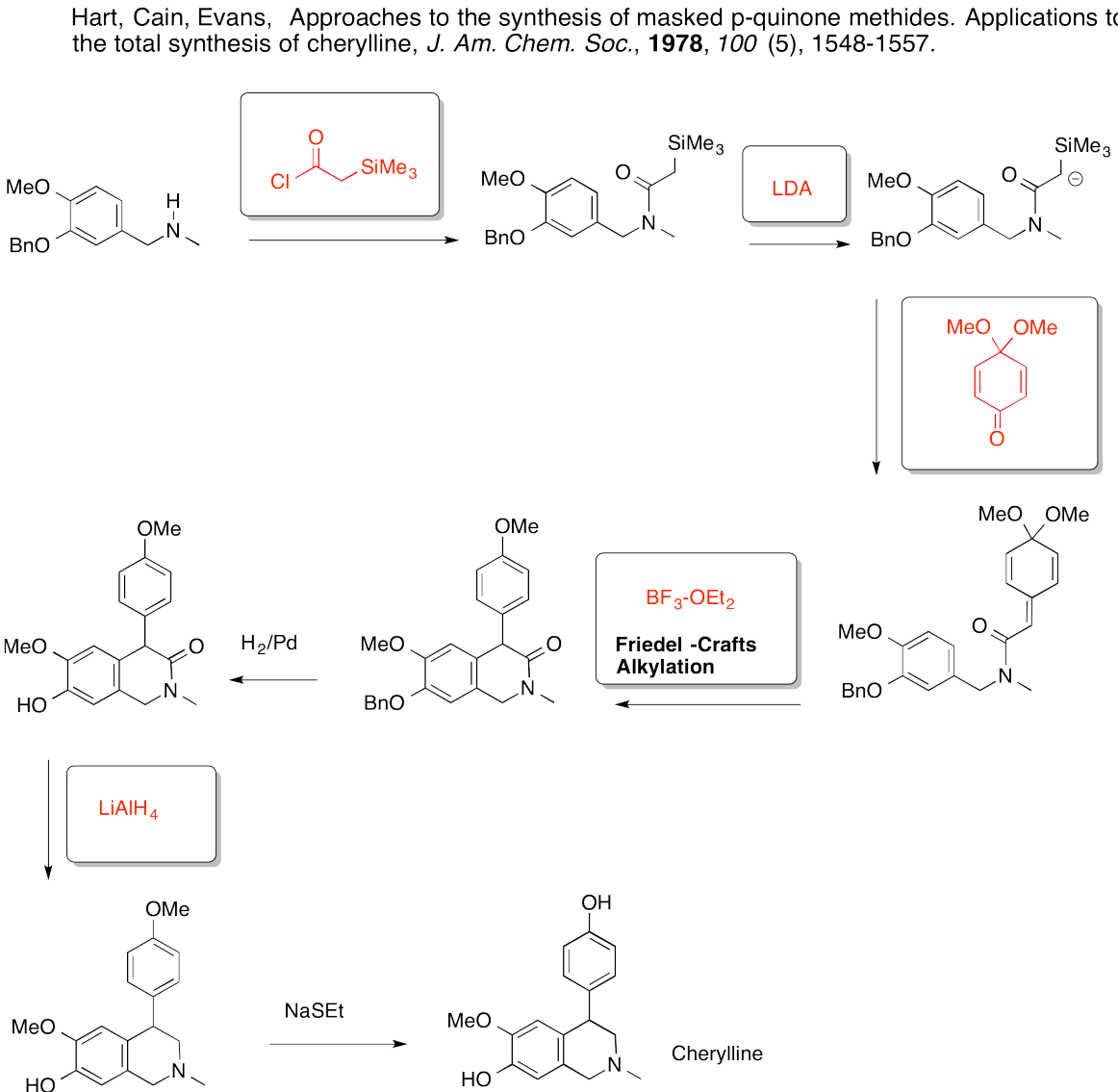
7.5: Directing Effects
- Page ID
- 189878

In addition to exerting an effect on the speed of reaction, substituents on the benzene ring also influence the regiochemistry of the reaction. That is, they control where the new substituent appears in the product.
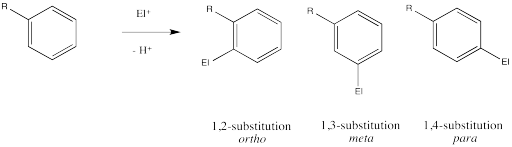
Remember, there are three different position on the bezene ring where a new substituent can attach, relative to the original substituent. Substitution could actually occur on five positions around the ring, but two pairs are related by symmetry. Isomerism in disubstituted benzenes can be described by numbering the substituents (1,2- etc) or by the relationships ortho-, meta- and para-. There are two positions ortho- to the initial substituent and two positions meta- to it.
Ingold and colleagues investigated the question of regiochemistry in nitration. They reported the following observations:
| Substitution patterns during nitration of benzene derivatives | |||
| R in C6H5R | % o- product | % m- product | % p- product |
| CH3 | 56 | 3 | 41 |
| F | 10 | 0 | 90 |
| Cl | 30 | 0 | 70 |
| Br | 38 | 0 | 62 |
| OH | 10 | 0 | 90 |
| CHO | 19 | 72 | 9 |
| CO2Et | 28 | 68 | 3 |
| CN | 17 | 81 | 2 |
| NO2 | 6 | 94 | 0 |
In looking at the table, you might see that there are two groups of substituents. One group reacts to make mixtures of ortho- and para- products. There may be different ratios of ortho- to para- and there may be small amounts of meta-, but don't get bogged down in the details right now. Focus on the bigger picture. Some groups are "ortho-/para-directors".
The other group reacts to makemostly meta-substituted products. There may be small amounts ofortho- and para- products, but don't worry about that. Focus on the bigger picture. Some groups are "meta-directors".
These regiochemical effects are very closely related to the activating and directing effects we have already seen. If we want to understand this data, we need to think about things like π-donation, π-acceptance, inductive effects and cation stability.
Show resonance structures for the cationic intermediate that results during nitration of toluene (methylbenzene). Explain why a mixture of ortho- and para- substitution results.
- Answer
-
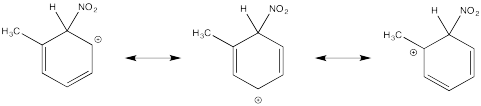
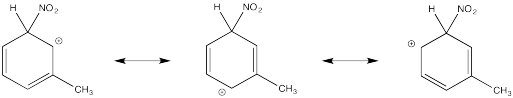
The tertiary cations that result during ortho- and meta- substitution offer extra stability, leading to preferential formation of these cations.
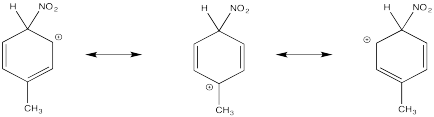
Show resonance structures for the cationic intermediate that results during nitration of chlorobenzene. Explain why a mixture of ortho- and para- substitution results.
- Answer
-
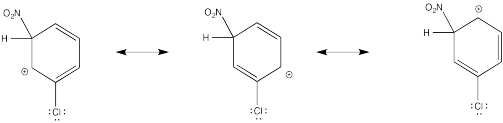
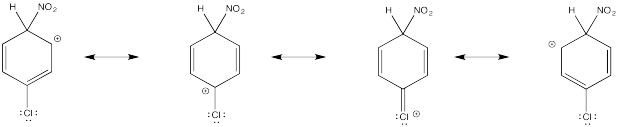
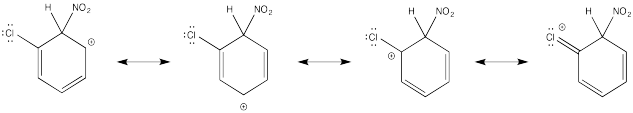
The π-donation that occurs in the cations arising from ortho- and meta- substitution results in extra stability, leading to preferential formation of these cations.
Show resonance structures for the cationic intermediate that results during nitration of acetophenone (C6H5COCH3). Explain why mostly meta- substitution results.
- Answer
-
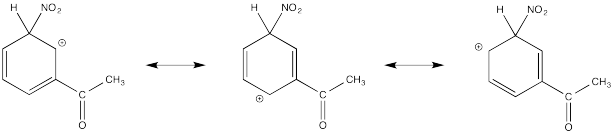
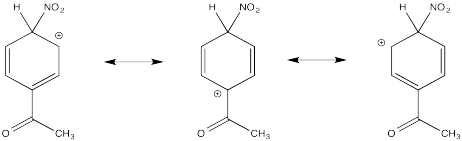
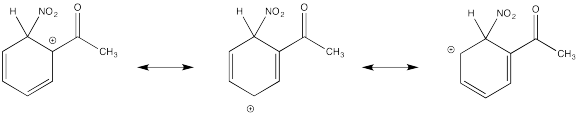
The cation directly adjacent to the carbonyl is destabilized by the electron withdrawing effect of the ketone. By default, the other intermediate is preferentially formed.
Show resonance structures for the cationic intermediate that results during nitration of acetanilide (C6H5NH(CO)CH3). Explain why a mixture of ortho- and para- substitution results.
- Answer
-
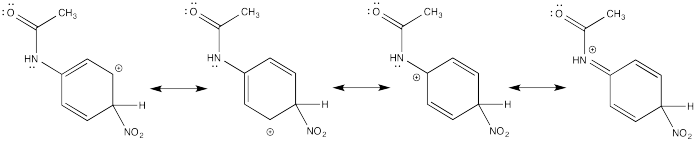
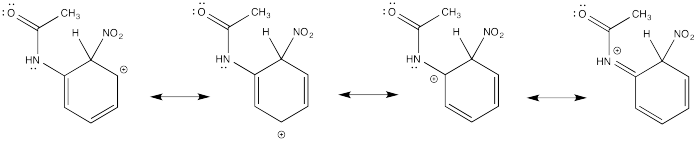
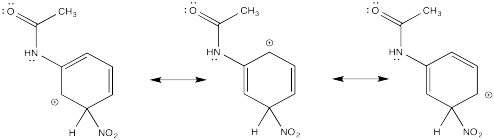
The π-donation that occurs in the cations arising from ortho- and meta- substitution results in extra stability, leading to preferential formation of these cations.
In general, we can divide these substituents into three groups:
- π-acceptors are meta- directors.
- π-donors are ortho-/para- directors.
- alkyls are ortho-/para- directors.
Note that, once again, we may have two competing effects in one substituent, such as a halogen. In halogens, although the net effect may be to slow the reaction down, that weak π-donation is still enough to tilt the balance of products in favor of ortho- and para- substitution.
For example, fluorine has a lone pair. It can be a π-donor.
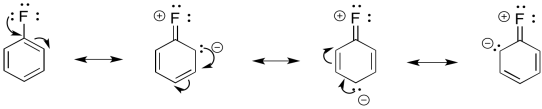
We could illustrate the effects with the following cartoon. It shows negative charge buildup, illustrated in red, on three of the carbons on the benzene ring.
Fluorine is also very electronegative. It can be an electron withdrawing group. Because it withdraws electrons through its sigma bond rather than through resonance effects, we think of it as "inductively" electron withdrawing. Nonetheless, a considerable amount of electron density from the benzene is attracted to the fluorine.
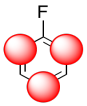
Again, we could illustrate the effects with a cartoon. It shows positive charge buildup, illustrated in blue, on the carbons that are closest to the fluorine. Remember, electrostatic interactions decrease rapidly with distance, so the farther the carbons are from the fluorine, the lower the effect that they will experience.
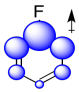
The sum of these effects can be illustrated in a composite cartoon that shows different amounts of positive or negative charge built up on different carbons.
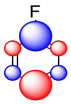
Thus, although fluorine is generally an electronegative and deactivating group, it may still manage to place some extraelectron density on particular carbons.
Whether something is overall activating or deactivating depends on a similar balance of factorsIn general, we can think about competing effects in terms of a see saw.
For an electronegative π-donor like fluorine or another halogen, the stronger those σ-withdrawing effects, the slower the reaction. It pulls more electron density away from the benzene than it puts back. As a result, the benzene is less nucleophilic than it would be if the halogen were not there.

On the other hand, the better the π-donation, the faster the reaction. Good π-donors include oxygen and nitrogen. They are just the right size to lend a lone pair to a neighbouring carbon in the benzene ring. Halogens, on the other hand, are relatively clumsy donors. Either they are just a little too electronegative to be useful, like fluorine, or else they are a little too big to share wtih carbon. That's the case with chlorine, bromine and iodine.
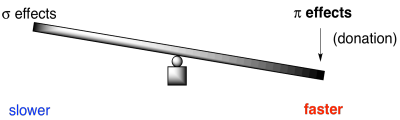
Remember, the same argumants are true in carbonyl chemistry. The general reactivity of carboxyloid derivatives depends on a similar balance between donating and withdrawing effects. As a result, esters and amides are relatively stable and unreactive, whereas chlorides are very reactive.
Of course, a π-acceptor would work in the opposite way. That would be true if the atom attached to the benzene is multiply bonded to another atom, especially if that other atom is something electronegative (an oxygen or a nitrogen).

Retrosynthetic Planning
Sometimes in synthetic work it is useful to look at a compound and imagine what it might be made from. This practice is used in industrial chemistry, when a researcher might be trying to decide on the most economical routed to make a particular compound. There may be readily available materials that a useful pharmaceuticla can be made from. By working backwards one step at a time, the researcher may more easily see different possibilities for starting materials. In a similar way, a biological chemist might be able to identify what biological precursors could lead to the formation of a particular compound in biology.
For example, by looking at the structure of m-bromonitrobenzene, you could imagine that the compound could be made by the bromination of nitrobenzene, but not by the nitration of bromobenzene. That's because nitro groups are meta-directors, but bromo-groups are ortho-, para-directors.
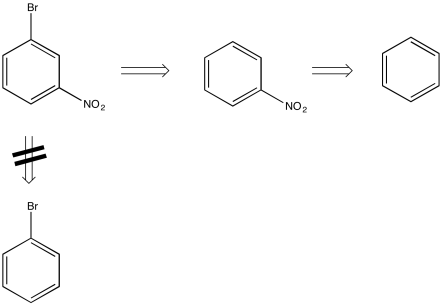
This method of working backward from the target compound is called "retrosynthetic analysis". By making a retrosynthetic plan, we can more efficiently arrive at the possible ways to make a specific compound.
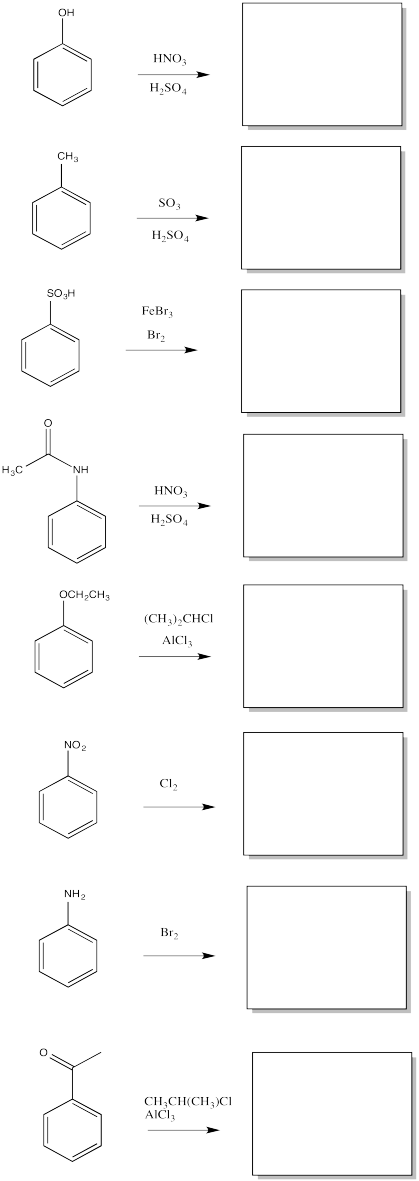
Fill in the major organic products of the following reactions.
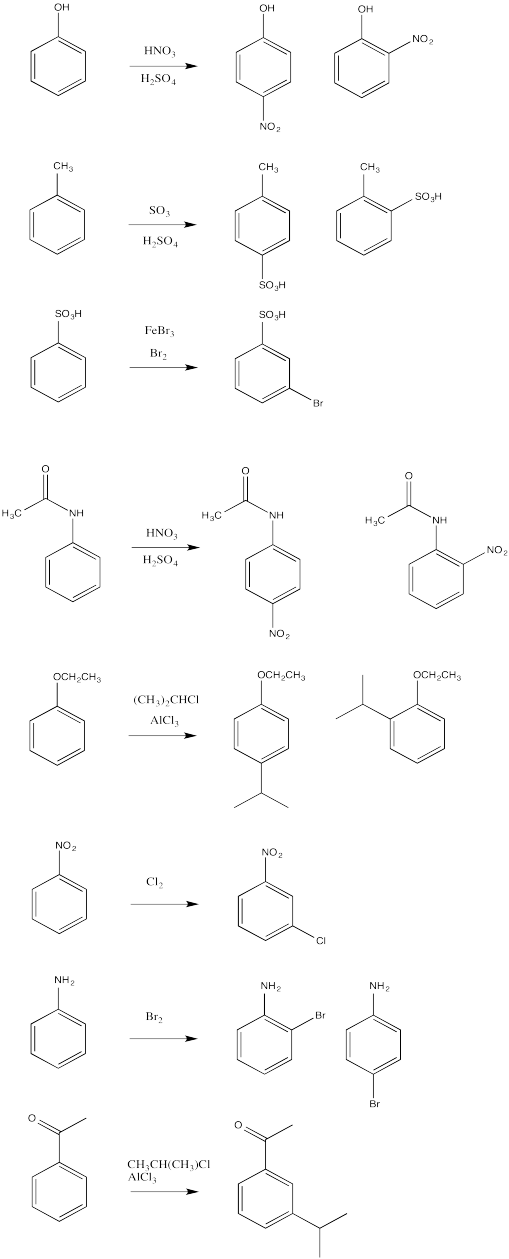
Answer-
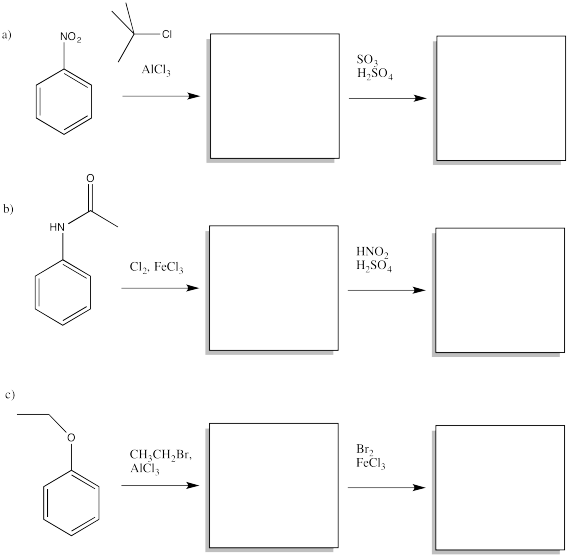
Fill in the starting materials and reagents needed to obtain the major product shown via electrophilic aromatic substitution.
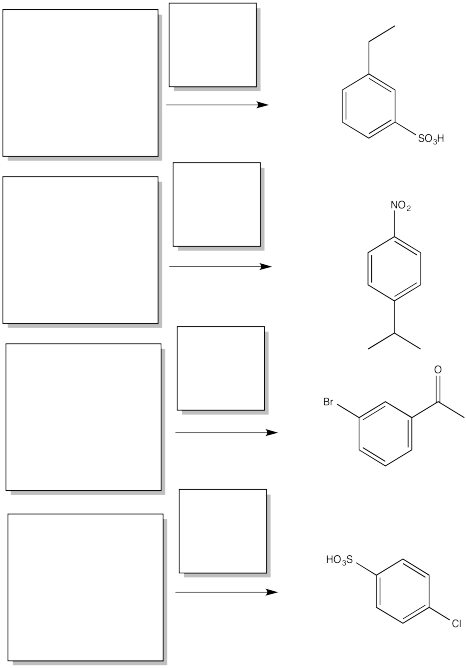
- Answer
-
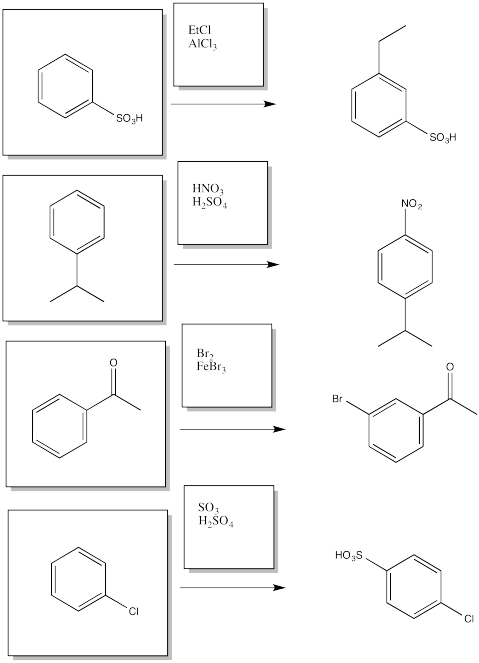
Given two different substituents on a benzene, there can sometimes be a conflict in predicting which substitution pattern will result. Generally, the group with the stronger activating effect wins out. Predict the major products of the following reactions.
- Answer
-
In cases leading to mixtures of ortho and para products, only one product was chosen, based on minimal steric interactions.
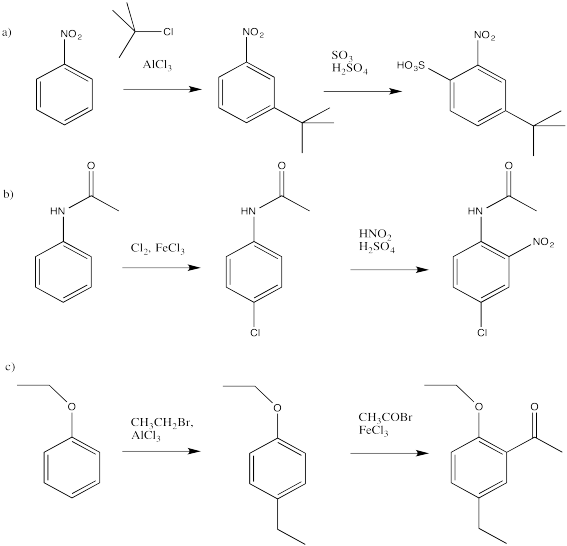
Problem AR5.8
- Answer
-
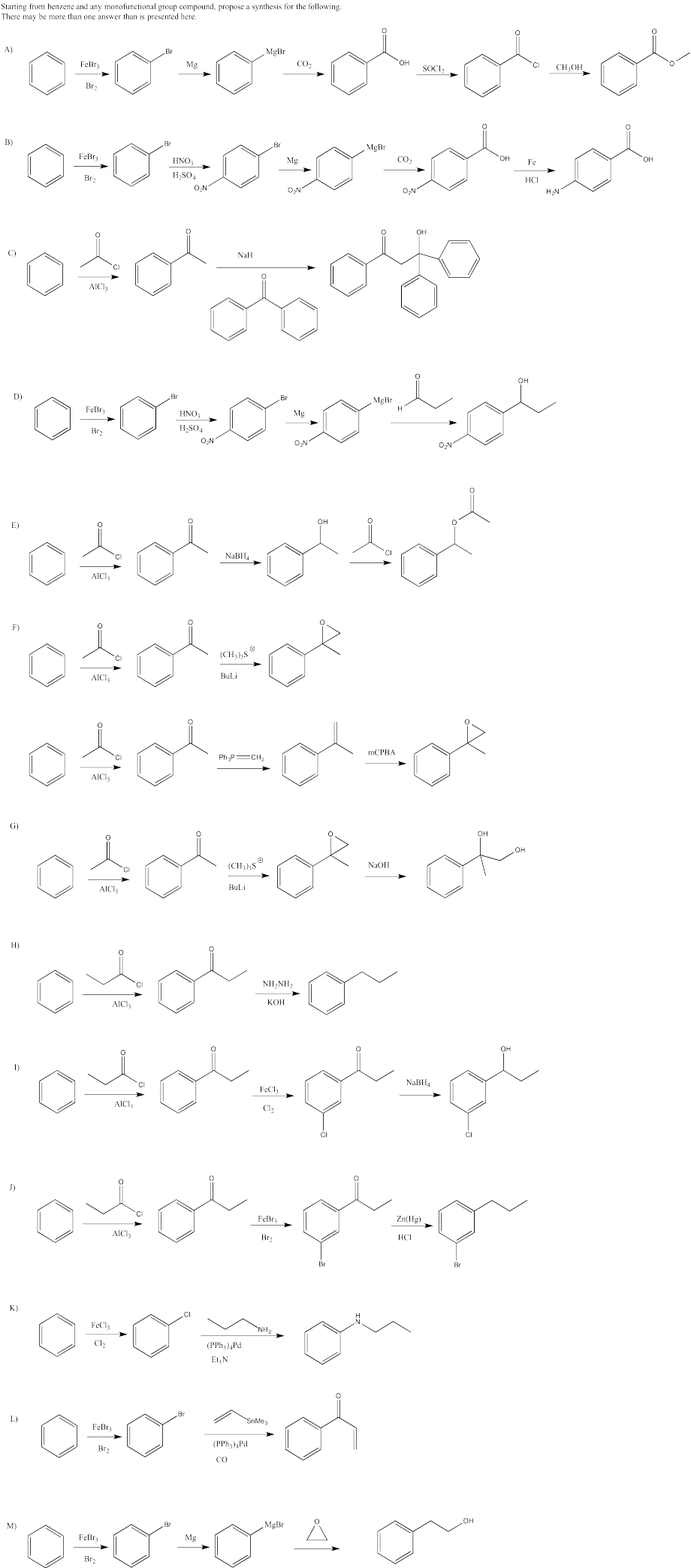
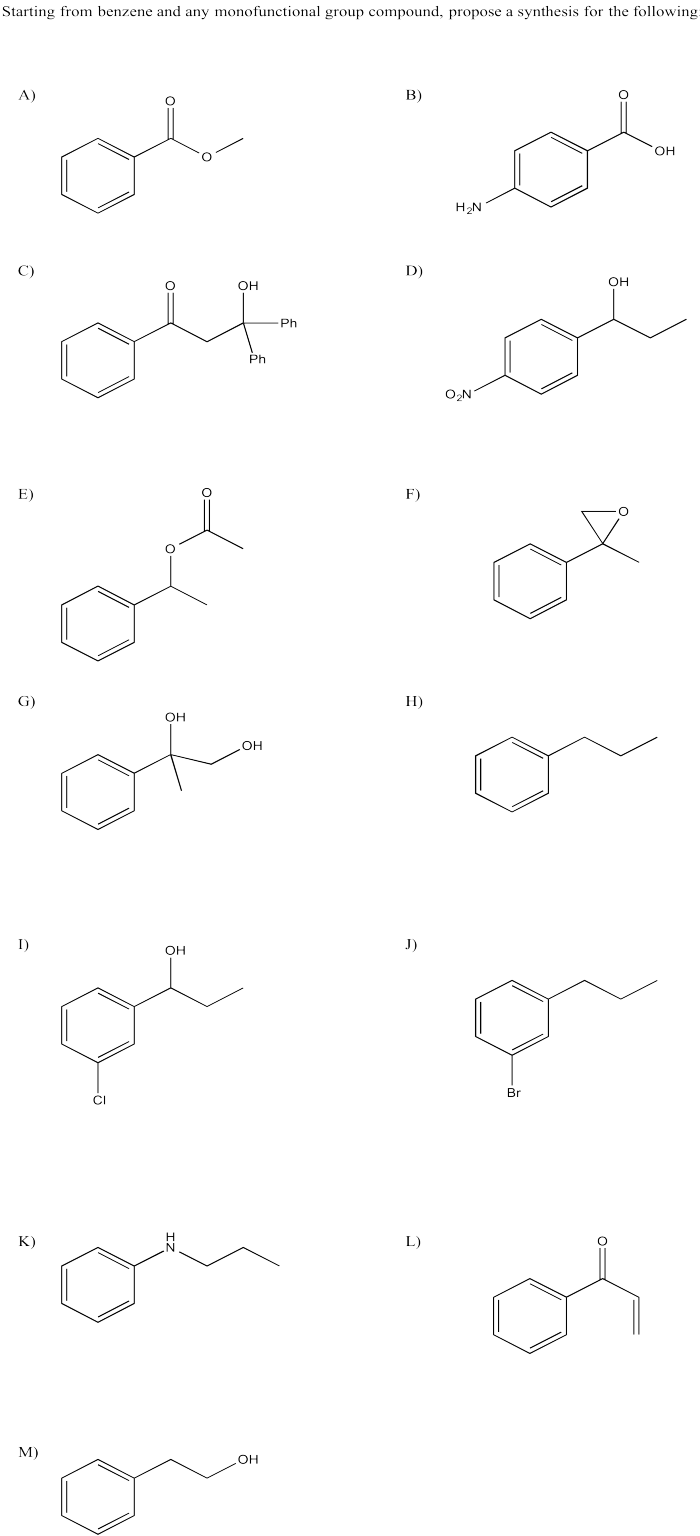
Provide a retrosynthetic plan for each of the following compounds, going back to benzene.
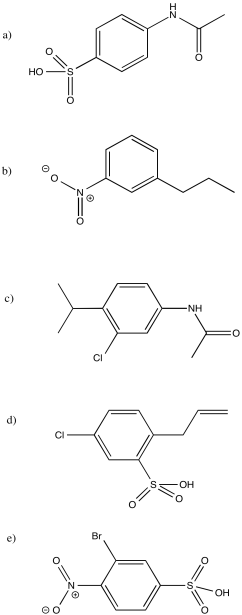
- Answer
-
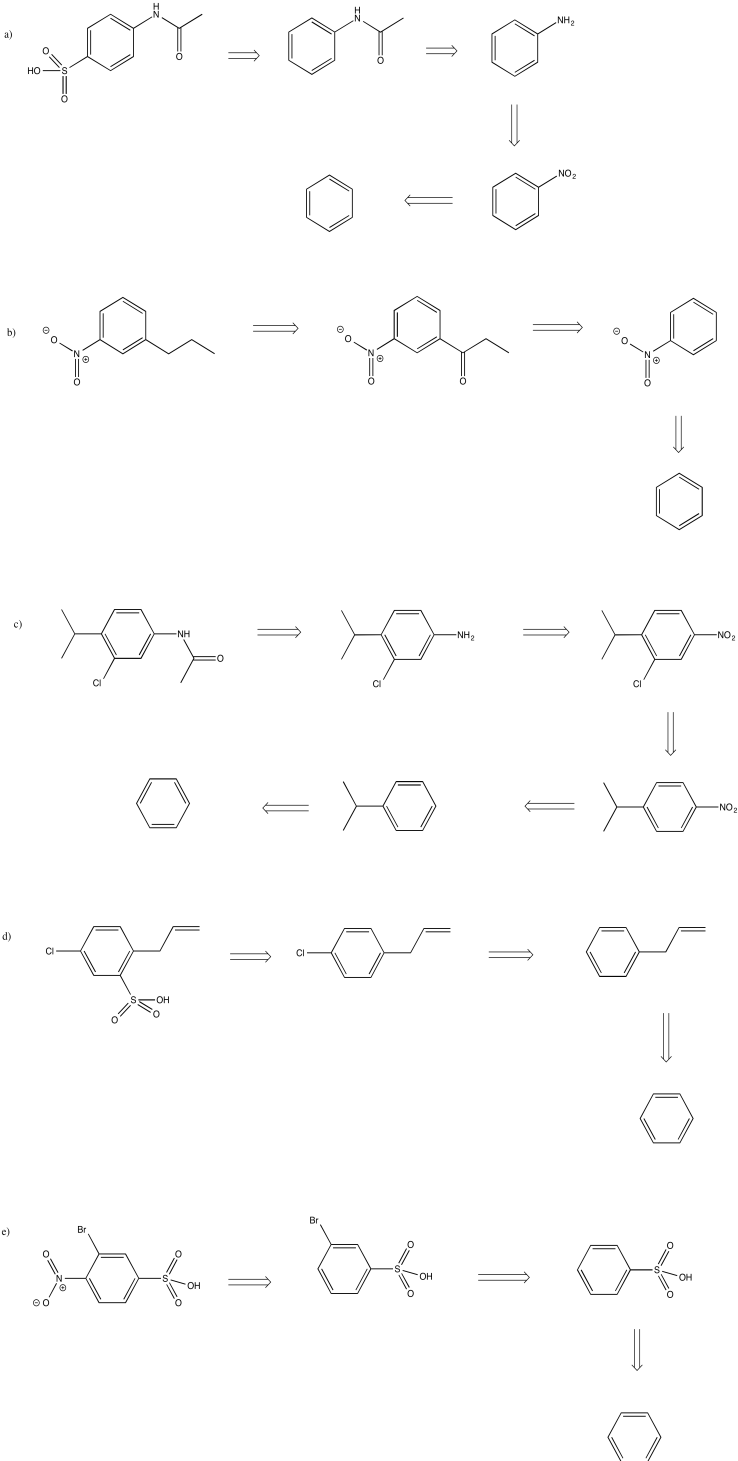
Turn the retrosynthetic plans from the previous problem into syntheses.
- Answer
-
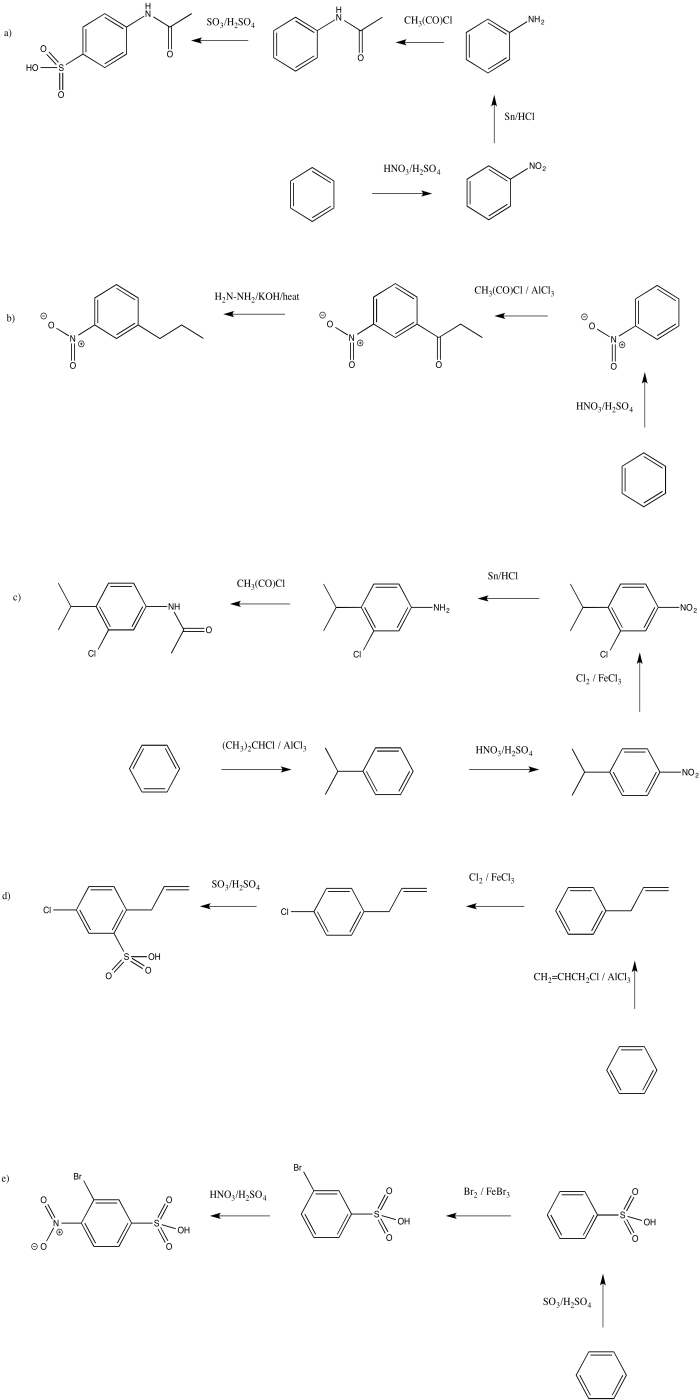
- Answer
-
Problem AR5.12.
- Answer
-