
10.3: Catalysis in Fatty Acid Synthesis
- Page ID
- 189983

After the last page, we would have you believe that fatty acids are constructed from acetyl coenzyme A units, each acting as consecutive nucleophiles, building the structure two carbons at a time.
There are a number of aspects of the biosynthesis that we didn't consider in that synthetic overview, but we are going to add some extra levels of understanding now. The most obvious difference is that the biosynthesis does not even involve acetyl coenzyme A, although it does involve a related thioester. Prior to the reaction, the initial acetyl coenzyme A electrophile is attached to an acyl carrier protein (ACP) via a cysteine residue on the protein.
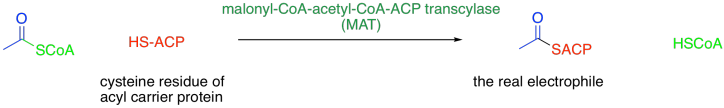
You can think of this as a regulatory step. The attachment of the acetyl group to the acyl carrier protein allows it to interact with the series of enzyme domains responsible for fatty acid biosynthesis.
This protein-attaching step is catalysed by malonyl-CoA-acetyl-CoA-ACP transcylase (everyone just calls it MAT). MAT uses a group transfer catalytic strategy, securing the acyl (or malonyl) group in place with a serine residue. The serine hydroxy group displaces the CoAS- thiolate from the acetyl, temporarily making it a regular ester. The serine is then displaced by a cysteine residue on the acyl carrier protein, or ACP. After that, the same sequence occurs with the malonyl group.
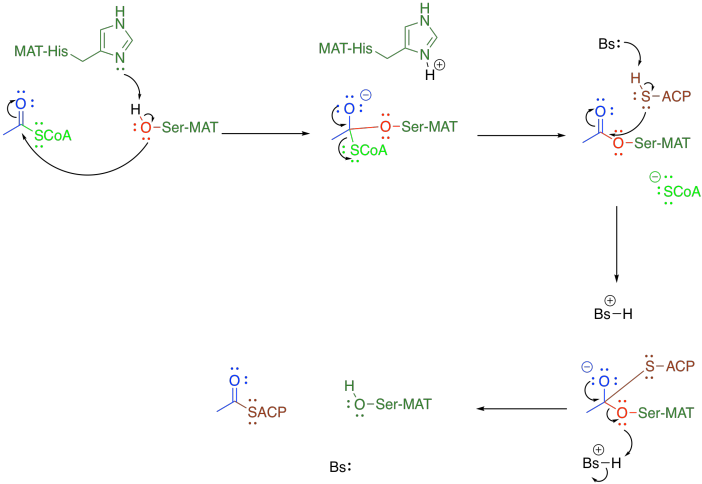
As in many such cases, the nucleophilicity of the key serine residue in MAT is enhanced through a hydrogen bond with a neighbouring histidine. This situation is pretty common in group transfer catalysis, especially in proteases. Frequently in these situations, a nearby aspartate or helps to in turn deprotonate the histidine (that's the so-called "catalytic triad" of ser-his-asp), but that part doesn't happen this time.
There is another big difference in the real biosynthetic route. In the Claisen condensation steps, the nucleophile does not really come from acetyl coenzyme A. Instead, it comes from malonyl coenzyme A. Acetyl coenzyme A and malonyl coenzyme A come together in a reaction catalysed by beta-ketoacyl-ACP synthase.

Notice that the malonyl group is also first attached to an acyl carrier protein, just like the acetyl group. That step is also carried out by MAT, and it happens in the same way.
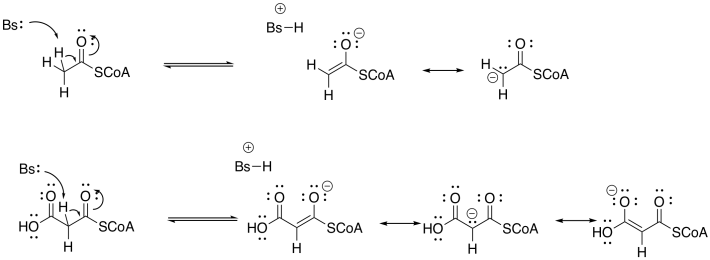
What is the point of using malonate thioester instead of acetate thioester for one of the components? It doesn't look very efficient; a molecule of carbon dioxide is lost, rather than just the four-carbon product. There are a couple of possibilities to consider here. First, the malonyl group is more acidic than the acetyl group, so it should be easier to convert into an enolate ion via removal of a proton.
Exercise \(\PageIndex{1}\)
Show why the malonyl ester would be deprotonated so much more easily than the acetyl ester.
- Answer
-
In the case of the malonyl enolate, there is additional delocalisation as demonstrated by resonance. This anion has extra stability.
Exercise \(\PageIndex{2}\)
If the reaction did happen this way, there would still be an extra CO2 in the structure after th Claisen reaction. Propose a mechanism for the Claisen reaction, and the loss of CO2 from the product.
- Answer
-
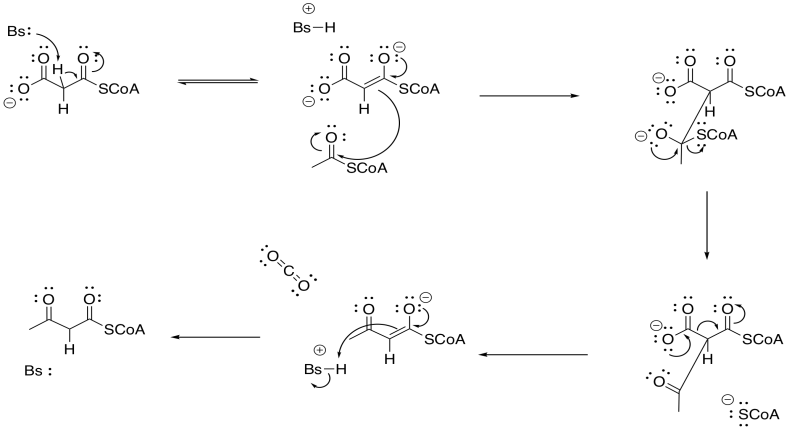
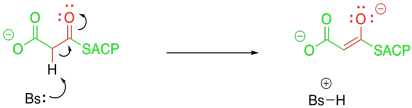
There are plenty of cases where this "malonate ester strategy" make reactions involving enolates much easier. Most biochemists think there is something different going on here, though. Rather than forming the enolate via proton removal, it is believed that the enolate forms via a decarboxylation reaction.

Decarboxylation reactions happen pretty easily if the CO2 group is beta to another carbonyl. That's because of the relative stability of that enolate anion. This decarboxylation reaction, therefore, accomplishes two things at once: it gets rid of the extra carbon in the malonyl group, and it generates exactly the enolate needed for the Claisen condensation.
There is an additional reason why the pathway employs the malonyl group. The formation of carbon dioxide is exothermic because of the formation of a strong C=O bond, as well as being entropically favored. Those factors help make the decarboxylation a driving force for the reaction.
The beta-ketoacyl-ACP synthase (or KS) also uses a group transfer strategy to hold the acetyl group in place. This time, a cysteine residue on KS displaces the thiolate of the acyl carrier protein.
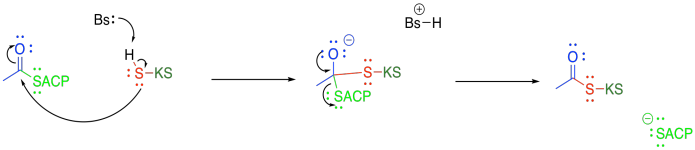
Once the acetyl group is tied down in the active site of KS, the malonyl-ACP enters the active site and undergoes decarboxylation, followed by Claisen condensation. The final part of the Claisen condensation releases the enzyme, completing the group transfer catalysis.
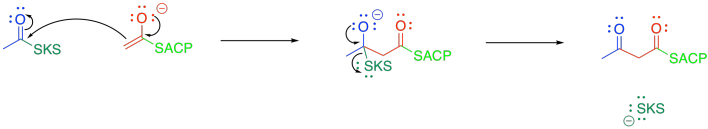
The subsequent steps aim to replace the beta-keto group with hydrogens. The first hydride is delivered with the help of beta-ketoacyl-ACP reductase, or KR.

It's a pretty standard mechanism. Frequently in enzymatic reactions, the delivery of a proton happens almost at the same time as the delivery of a nucleophile to a carbonyl. In this case, the nucleophile is a hydride ion delivered by NADPH. The proton comes from some amino acid residue in a protonated state; lysine has been used in the illustration as an example.
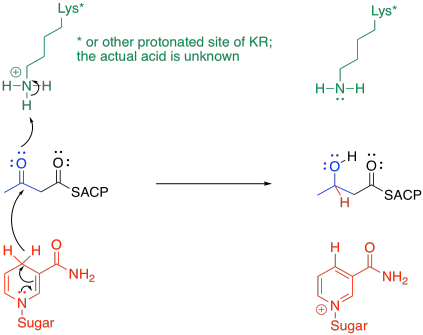
Elimination of water is promoted by beta-hydroxyacyl-ACP dehydratase, or DH.

This looks like a tricky reaction. It depends once again on the ease of removing a proton from that alpha position. That's not terribly difficult, because a lone pair in the alpha position is stabilised by resonance. The other problem is pushing off the hydroxy group. That isn't a very good leaving group, so it poses some problem. However, it is thought that the histidine that deprotonates the alpha position then delivers the proton to the hydroxyl group. As a result, the hydroxyl group can leave as a water molecule.
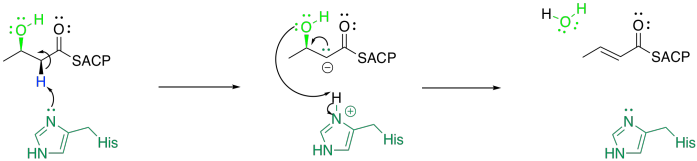
This pathway for an elimination reaction (the loss of a proton from one carbon and a leaving group from the next) is called an E1CB reaction. The CB in this acronym stands for the conjugate base that is formed after the deprotonation.
The final portion of the cycle is another hydride addition, this time carried out by beta-enoyl-ACP reductase (ER).

The mechanism is pretty similar to the first hydride addition. Once again, the exact nature of the proton donor is a little unclear, but the hydride donor is another NADPH molecule. This time, the NADPH must be positioned so that it delivers the hydride to the 1,4-position rather than to the carbonyl.
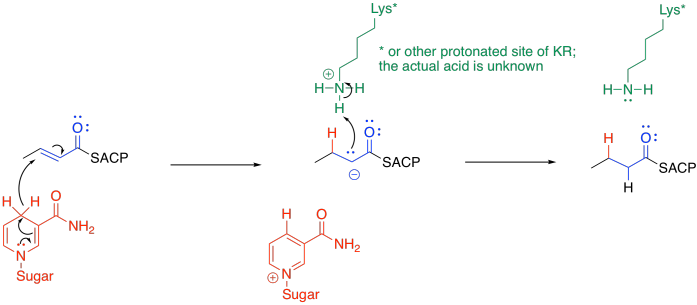
The final product of this cycle could be released or it could be sent back for another turn of the cycle, extending the fatty acid chain by two more carbons.