
6.3: Strategies in Enzyme Catalysis
- Page ID
- 189955

Once a substrate has been bound, it is the enzyme's job to quickly transform the substrate into product. The enzyme does so by carrying the substrate over a catalytic pathway. In a catalytic pathway, the reaction takes a different course than it would on its own. Sometimes the catalytic pathway is longer, involving additional steps, but because the energetic terrain is easier to traverse than in the un-catalyzed process, the catalytic reaction actually takes much less time.
Enzymes have a range of structures and reaction properties, so there are a wide number of different reactions they can catalyze. Nevertheless, there are a few common strategies displayed in catalytic reactions that are useful to know.
Approximation
When we make an approximation, we are getting close to the answer. If someone asks us what time it is, and it is 3:02 pm, we probably tell them it's about three o'clock. That's close enough.
In enzyme catalysis, approximation means getting things close to each other. If we have a substrate that is going to react with something, the enzyme can bind the substrate in such a way as to get the substrate in close proximity to the reactant. Sometimes, but not always, that might mean binding two substrates, so that they can more easily react with each other. If you want to meet your friend, it's a much better idea to say, let's meet at Cafe Santropol at 7:00, rather than wander the streets of Montreal, along with four million other people, hoping to bump into your friend. The reactant and substrate are much more likely to encounter each other in the protected confines of the enzyme than they are floating around in the wilderness of the cell.
- In approximation, two substrates are held close together within the enzyme
- This proximity makes them react together more easily
Sometimes, this idea can be a little more subtle. Imagine that the reactant is the enzyme itself, so that just by binding the single substrate, the reaction is already much more likely to occur. The substrate may be held in such a way that it is already in close proximity to amino acid side chains that will work on it and transform it into a new molecule.
- In approximation, the substrate is held in position so that subsequent reaction is much more likely
Approximation is really an entropy factor. By binding the substrate, we can limit its degrees of freedom, restricting its location or even its orientation so that there is no way the reactant can miss its intended target.
Of course, the substrates still have to find the active site of the enzyme. Sometimes, that task is aided by sticky surfaces on the enzyme; there can be groups on the surface of the enzyme that can interact with substrates, so that the substrates are less likely to drift away after a random collision with the enzyme. With its movement thus restricted, the substrate is more likely to move into the active site than drift off through the cell once more.
Although we are discussing enzymes in the cell, other kinds of catalysis make use of approximation as well. For example, transition metal catalysis often makes use of solid chunks of metal to catalyze the reactions of gaseous vapors. The surface of the metal gives the gas-phase molecules a place to bind, giving them a place to gather, rather than wandering around in three dimensions in the gas phase. Once they are together in one place, they are more likely to react with each other or with additional species on the surface of the metal.
Acid-Base Catalysis
Acid-base catalysis is a very common phenomenon. So many reactions involve the addition or removal of protons, especially the carbonyl reactions that are so prevalent in biochemical pathways, that proton donors and acceptors become key players. Acidic and basic side chains of the amino acids in the protein naturally fill these roles.
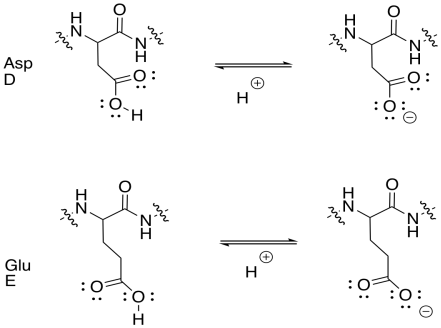
Which amino acids are thought of as having "acidic" side chains?
- Answer
-
Aspartic acid (abbreviations Asp or D) and glutamic acid (abbreviations Glu or E).
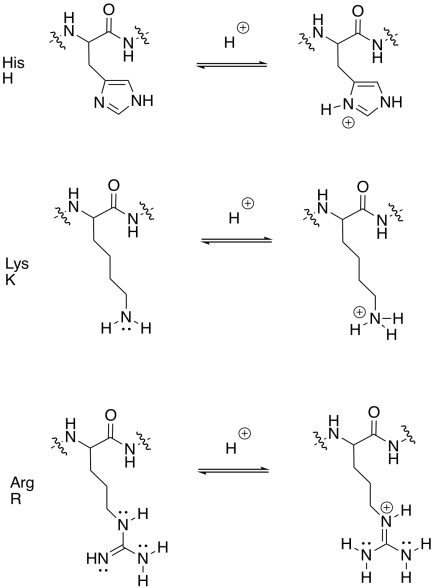
Which amino acids are thought of as having "basic" side chains?
- Answer
-
Histidine (abbreviations His or H), lysine (abbreviations Lys or K) and arginine (abbreviations Arg or R).
Draw the acidic amino acid side chains in both their acidic and conjugate basic forms.
- Answer
-
Draw the basic amino acid side chains in both their basic and conjugate acidic forms.
- Answer
-
Acid-base catalysis can provide mechanistic advantages by rapidly enhancing the electrophilicity of a molecule. Any carbonyl compound is electrophilic, but if it gets protonated, the overall positive charge makes the carbonyl even more electrophilic.

Alternatively, acid-base catalysis might increase the nucleophilicity of a molecule. Any alcohol is nucleophilic, because it has lone pairs. However, if its proton is removed, it becomes even more nucleophilic, because of the overall negative charge.
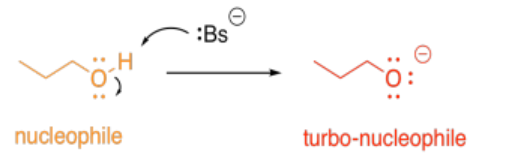
So, in the most straightforward case, adding a proton might accelerate a reaction involving an electrophile. Removing a proton might accelerate a reaction involving a nucleophile.
- Acid/base catalysis involves rapid proton shuttling
- Acidic side chains can activate electrophiles
- Basic side chains can activate nucleophiles
There are other variations on this approach. For example, consider a keto-enol tautomerism. We think of ketones classically as electrophiles, but their enol isomer is easily accessible in general, and the enol form is an excellent nucleophile. As part of a series of reaction, rapid conversion of a ketone into an enol might be a key step.

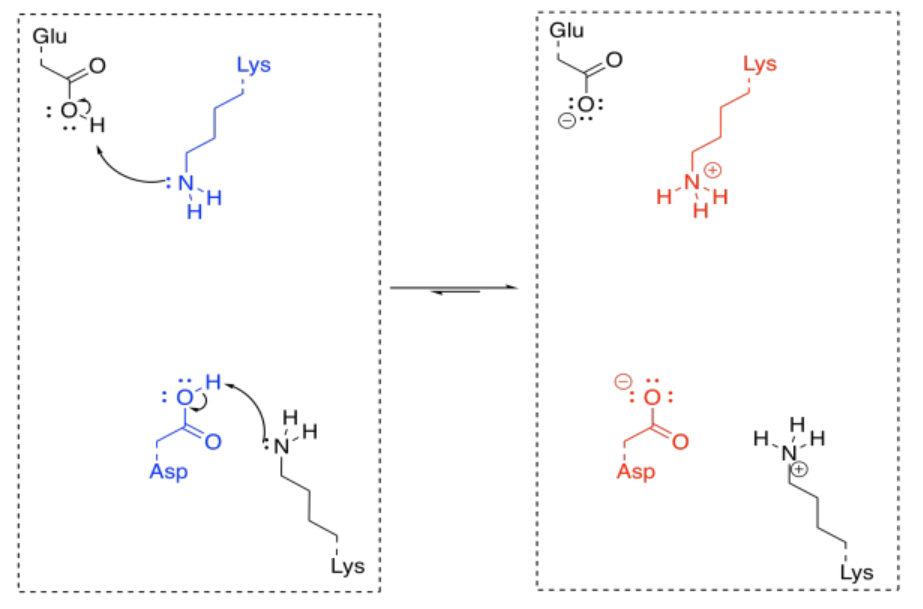
That's really a couple of different steps; you have to add a proton to the carbonyl oxygen, and then you have to take a proton away from the alpha position. You could do it the other way around, but it would still be two steps. Fortunately, enzymes have lots of acidic and basic sites, so there may be sources and sinks of protons that are readily available for a molecule bound in an enzyme. Furthermore, the molecule may be bound in such a way that these proton transfer steps are essentially simultaneous.
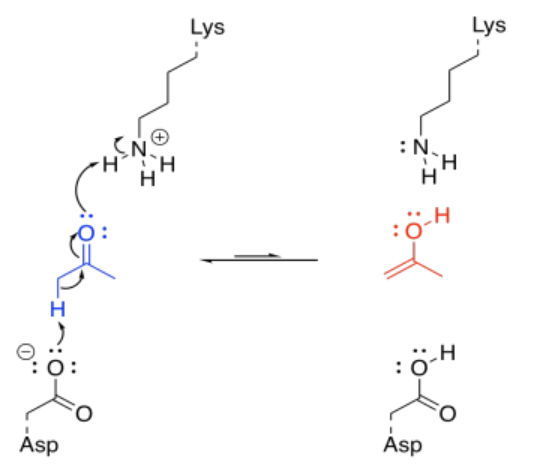
Under normal circumstances, this looks like three different molecules reacting together; that's basically impossible under normal circumstances. But if the lysine and the aspartate are part of the same enzyme, that reduces the problem to an interaction between two molecules; that happens all the time. If the ketone is already bound in the enzyme, the difficulty is reduced even further.
In order to make the reaction catalytic, the enzyme has to be regenerated. Again, that isn't a problem for enzymes, because of the number of acidic and basic sites available. A key site that is missing a crucial proton can pick one up from another amino acid side chain sitting nearby. Protonation states get reshuffled pretty quickly, and soon the enzyme is ready to go again.
Acid-Base Catalysis: Metal Ion Catalysis
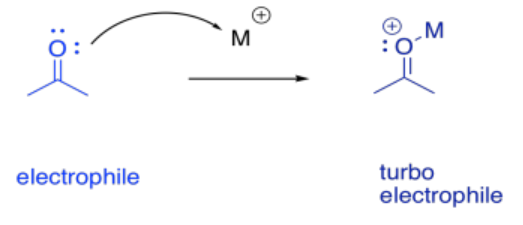
Metal ion catalysis can often be thought of as a special case of acid-base catalysis. With metal ions, we get Lewis acid catalysis. Lewis acid catalysis can accelerate reactions in a couple of different ways, in close analogy with general acid/base catalysis.
It sometimes helps to think of a metal as a great, big proton. That's an oversimplification, as we'll see in a moment. Nevertheless, it can be useful to keep the analogy in mind. When a compound binds to a metal ion, the effect can be similar to binding a proton. The compound has just donated a pair of electrons elsewhere (to the metal ion or to the proton), and so the compound suddenly looks electron-deficitient. It has enhanced electrophilicity.
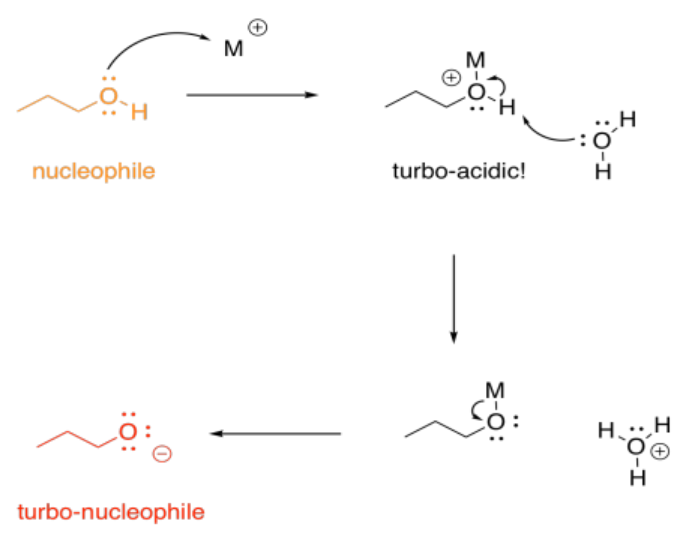
On the other hand, we don't usually think of protonation as causing increased basicity; that would be completely backwards. With metals, though, that can happen, indirectly. If an alcohol, for example, donates a lone pair to a metal, the oxygen becomes positively charged. It becomes much more acidic. Suddenly, the alcohol can be deprotonated by a very, very weak base, such as water. That leads to formation of an alkoxide ion, which is much more nucleophilic than the original alcohol. Water would really never take a proton from an alcohol, but it can do it once the oxygen has a positive charge.
Metal ions can play a number of other roles in catalysis, but that's enough to get an idea of just some of the ways in which they might be useful. To learn more, we would have to explore more transition metal reactivity, including the ability of metals to donate and accept individual electrons.
Group Transfer
Group transfer, also called covalent catalysis, has something in common with approximation. It limits the degrees of freedom involved in a key step of the pathway to product. Rather than just binding two substrates near each other in the active site, or holding a substrate in a specific orientation, group transfer involves a reaction between the substrate and the enzyme such that the substrate becomes attached to the enzyme. It is attached via a covalent bond.
In a sense, like approximation, this mechanism limits the problem of two substrates bumping into each other -- an unlikely prospect in the roliing sea of the cell -- to two things coming together within the limited space of the active site. Of course, any time a molecule wanders into an active site, there is a chance it could wander off again; but that isn't true if you fasten it down. That's what group transfer does. It fastens one substrate to the enzyme, so that a second substrate is more likely to react with it.
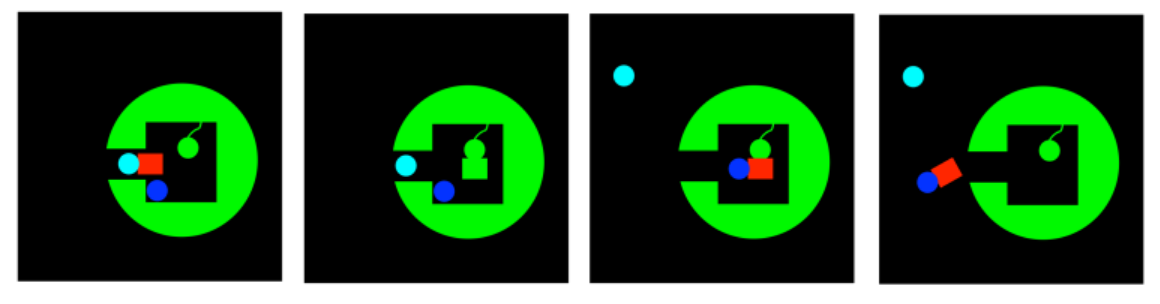
That's not all. Sometimes when the substrate, or a group forming the crucial part of the substrate, is transferred to a side chain on the enzyme, it becomes modified in such a way as to become more reactive. For example, an aldehyde may be electrophilic to begin with. However, if it becomes attached to a lysine residue, it may be transformed into an imine. Now, an imine may seem less electrophilic than an aldehyde, because nitrogen is less electronegative than oxygen. However that same feature also makes nitrogen much more basic than oxygen. As a result, imines are very likely to rest in a protonated state; aldehydes are much less likely to be protonated. That protonated iminium ion, of course, is much more electrophilic than an aldehyde, because the iminium has a full positive charge.
Group transfer may even result in complete reversal of reactivity in the substrate. Again, in the case in which an aldehyde becomes tethered to a lysine residue, the aldehyde may be transformed into an enamine. The difference between forming an enamine and an imine is just the difference in which of the protons is removed. In an enzyme, that decision is easily settled by the positioning of the amino acid side chain that removes the proton. In one enzyme, this side chain removes the nearest proton, leading to an imine. In another enzyme, another side chain, situated in a different place, removes the nearest proton, leading to an enamine.
Of course, the consequence of forming an enamine rather than an imine is much like having an enolate or enol rather than an aldehyde. The former is nucleophilic, even though the latter is electrophilic. So, group transfer can even flip something from an electrophile into a nucleophile.
A very different example that is also pretty common involves a set of amino acid side chains called "the catalytic triad". These three groups -- aspartate, histidine, and serine -- are frequently observed in group transfer catalysis. With the catalytic triad, it is the serine that acts as the nucleophile, rather than a lysine. As a result, it is the serine that becomes bonded to the substrate. The role of the aspartate and histidine is to act as a proton relay, helping to activate the serine so that it becomes a more effective nucleophile.
Transition State Stabilization
We think of enzymes as being perfectly suited to bind a particular substrate, but that may not be the case at all. If you think about it, an enzyme that perfectly matches its substrate would pick up its substrate, snap it into place and... that would be all. Nothing else would ever happen. The substrate would sit there forever. Why mess with perfection?
Instead, it is thought that, at least in some cases, the enzyme is ideally suited to bind something a little further down the path. The idea is that the substrate will change shape as it goes through the reaction, and if the enzyme actually better fits the shape of something else to come, it will exert pressure on the substrate to go ahead and react. Once the substrate gets to the right shape, it will be rewarded with perfect binding.
In the extreme case, we might think about the enzyme being a perfect match for the product. That way, the enzyme could coax the reaction along, and the strong binding interaction between enzyme and product would pull the reaction through its equilibrium all the way to the product side.
Well, if you think about it, that wouldn't work very well, either. Once the product forms, it would bind perfectly with the enzyme, which would never let it go. What good is it if the product is made but never gets released into the cell? The whole point of enzymatic catalysis is to quickly make things that the cell desperately needs; without these things, the cell will die.
But there is one kind of structure that could bind the enzyme perfectly, and we wouldn't have to worry about it getting stuck. It's a transition state. A transition state is an inherently unnatural structure; the molecule can't stay that way for long. It's just passing through on its way from one structure to another, and the transition state is the awkward, gangly phase in between, that horrid thing we glimpse in the dark when it thinks we aren't looking.
So if we stabilize that thing, then the substrate is pushed onward, a little further, and just when it reaches the perfect point, it finds it's gone too far. It must collapse. It's like Wile E. Coyote sublimely achieving his goal just as he passes over the cliff's edge.
Transition State Stabilization: Distortion
If transition state stabilization is the carrot, then distortion is the stick. Transition state stabilization coaxes the substrate further on; distortion pushes and prods until the substrate can take it no longer.
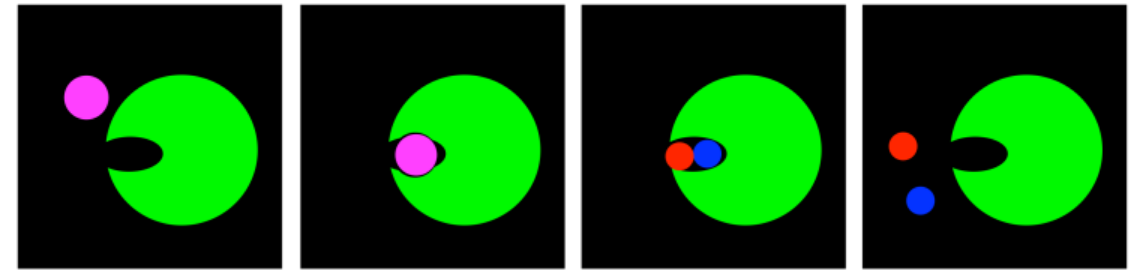
In distortion, the substrate finds itself in a binding site in which its shape is not quite right. Its geometry is subtly squeezed. The substrate itself is destabilized, whereas forming the product, or the transition state leading to the product, actually relieves that strain.
A good example is an atom that is supposed to be trigonal planar, but finds itself sitting in a place where it can't quite lay flat. Instead, it finds intermolecular interactions that are pushing it into another shape; maybe tetrahedral would be a better fit.
If a tetrahedral intermediate lies ahead, then by the time we reach the transition state, the geometry of that atom may actually look more like a tetrahedron than a trigonal plane. As a result, those intermolecular forces are distorting the otherwise stable structure of the substrate and bending towards the transition state.