
6.2: Reaction Concepts
- Page ID
- 112511

Overarching Concepts
Reaction types are significant to all fields of chemistry because they are what characterize any transformation from starting materials to products. In the pantheon of reactions, we tend to focus on functional group creation and atom transformation, both of which can be accomplished through the menu of reaction types shown below:
- Rearrangement (RAR)
- Addition
- Substitution
- Elimination
- Pericyclic reaction
- Ox/Red
- Reaction design
- Atom economy
The above six (6) general reaction categories simplify the panoply of reactivity paradigms in the discipline of chemistry. We can nearly list every pertinent reaction within one or more of the listings shown. Indeed, even acid/base chemistry can fall under Addition or Elimination reactions based on their precise reactivity patterns. We will delve deeply into each type in the following sections.
Rearrangements (RARs)
A rearrangement reaction covers a broad swath of chemistry which is principally characterized by the rearrangement of the skeletal configuration of a molecular system to yield a structural isomer (a swapped-out form of the original structure while retaining the original atoms). Oftentimes, a substituent moves from one backbone atom to another backbone atom. In the classical organic chemistry example shown below, we witness the Wagner-Meerwein RAR.
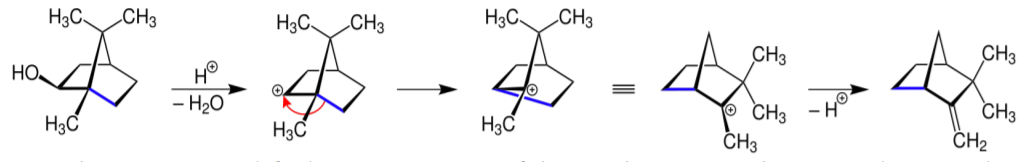
A rearrangement is not exactly well depicted by discrete electron transfers shown by curly arrows. The actual mechanism of alkyl groups moving in Figure \(\PageIndex{1}\) involves a fluid transfer of the alkyl group along a bond that is not described by simple ionic bond-breaking and reformation. Explanation by orbital interactions provides a better approximation although it is possible to draw the curved arrows for a sequence of discrete electron transfers to give the same result as a RAR reaction.
There are other types of RARs besides the chemically induced one shown above: we can have thermal or photo-induced as well. In these latter cases, heat or light can act as the triggers to actuate the transformation.
Thermal RARs
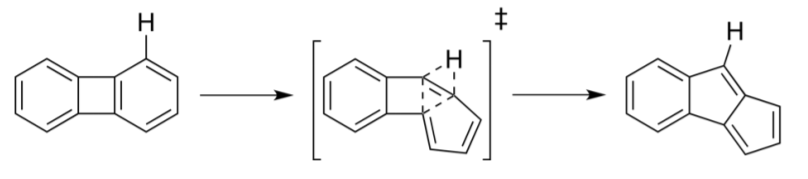
The isomerization of unsubstituted azulene to naphthalene was the first reported and hence most studied thermal transformation for an aromatic hydrocarbon. Many mechanisms have been suggested, yet not a one has been unequivocally confirmed.
Five mechanisms were proposed: reversible ring-closure, shown above, a norcaradiene- vinylidene, diradical, methylene walk, and a spiran mechanism. The reversible ring- closure mechanism is inaccurate despite its overall scientific appeal and evidence, and as such, multiple reaction pathways were deemed occurring simultaneously. This idea was widely accepted. It was thought that at high temperatures one mechanism would be energetically favored although energetic studies displayed similar activation energies for all mechanisms [1].
Four mechanisms for thermal isomerizations are warranted: dyotropic, diradical, and two benzene ring contractions. A 1,2-carbon shift to a carbene preceding a 1,2-hydrogen shift, and a 1-2- hydrogen shift to a carbene followed by a 1,2-carbon shift. The dyotropic mechanism shows the 1,2-shift displayed in Figure \(\PageIndex{2}\) .
Photoinduced RARs
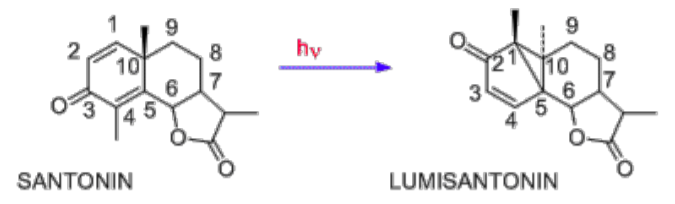
The mechanisms of organic photochemical reactions are a treasure trove for photochemists and physicists because they provide tremendous insight into fundamental electronic behavior as a function of energy-matter interactions. The absorption of ultraviolet light by organics may lead to a number of reactions. Over the last century, an immense quantity of photochemical reactions have become unearthed; it has been found that reactions that are unlikely in ground-states are accessible in electronic excited-state configurations. One of the earliest photochemical studies was on the natural product santonin. Ciamician (1857-1922) observed that under sunlight exposure, santonin gave several photoproducts. The structure of santonin (molecule on left in Figure 6-3) was first described by Clemo and Hayworth in 1929 whose initial photoproduct is lumisantonin. As depicted, the photoreaction involves a C-3 carbonyl group movement to C-2, the C-4 methyl has moved to C-1, and the C-10 carbon has been inverted.
This reaction is named after the German chemist Ernst Otto Beckmann (1853–1923). It is representative of an acid-catalyzed rearrangement of an oxime to an amide (when the oximes are cyclic, they yield “lactams”).
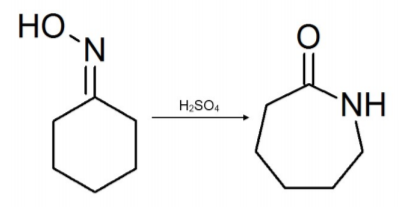
Caprolactam, in this example, is a very important chemical because it is the feedstock in the production of Nylon 6, a material used extensively in the manufacture of textiles, clothes, carpets, cushions, bulletproof vests, etc. The reaction includes acetic acid, hydrochloric acid, sulfuric acid, and acetic anhydride, to catalyze the rearrangement. Sulfuric acid is the most commonly used acid for commercial lactam production because it forms ammonium sulfate (a common agricultural fertilizer) as a by-product when neutralized with ammonia.
In general, rearrangements tend to have no waste of atoms because they are a type of isomerization. They are hence very atom economical and hence very efficient modes of chemical transformations. At this point, we will delve into a series of rearrangements to further illustrate the point.
Addition Reactions
This type of reaction occurs when in general two molecules covalently bind to form a larger one referred to as the adduct. These reactions are limited to chemical compounds that have multiple bonds, such as molecules with carbon–carbon double bonds (alkenes), triple bonds (alkynes) hetero double bonds like carbonyl (C=O) groups, or imine (C=N) groups. An addition reaction is typically the reverse of an elimination reaction. For example, the hydration of an alkene to an alcohol is the reverse of dehydration which leads to an oxidative product (alkene). Electrophilic and nucleophilic additions are the main types of polar addition. The non-polar additions can be free radical and cycloaddition.
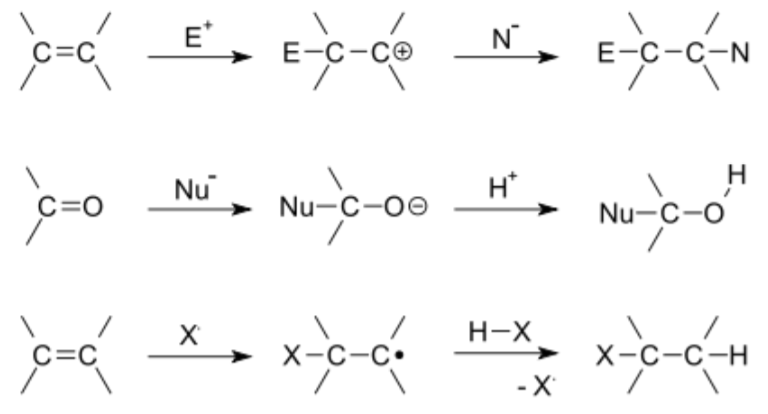
In general, addition reactions are very common because they are simple, very powerful, robust, and preserve/conserve atom economy during the reaction sequence (think of the mathematical operation of addition – all numbers sum!). Thus, in general, no additional by-products are generated allowing for a clean and straight forward reaction scheme that in the final analysis may require little to no purification steps.
Substitution Reactions
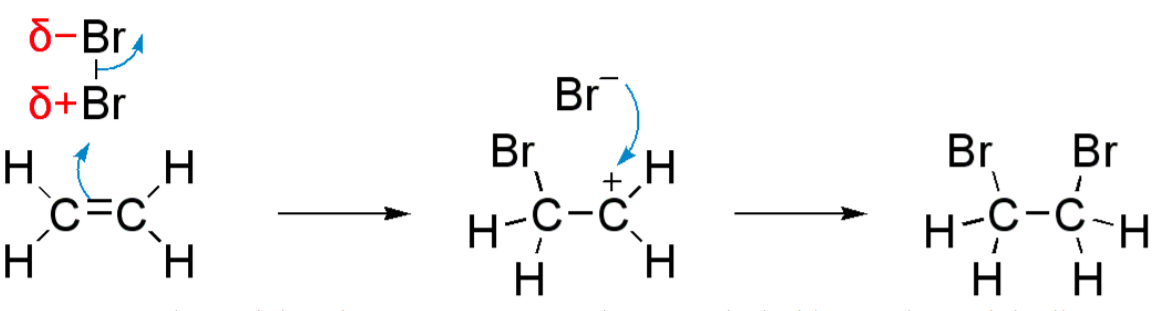
These types of reactions are also known as single displacement reaction or single substitution reaction). One functional group is replaced by a more reactive functional group or else it would not happen (the reverse would be faster or more thermodynamically stable). Substitution reactions are classified either electrophilic or nucleophilic depending upon the reagents. A good example of a substitution reaction is halogenation. When chlorine gas (Cl-Cl) is irradiated with the appropriate light energy, several of the molecules photolyze into two chlorine radicals (Cl·) whose electrons are very nucleophilic. In Figure 6-6, one of radicals can rupture (homolytically) a C-H bond through abstraction of one of the equivalent protons on methane to form electrically neutral H-Cl. The other radical reforms a covalent bond with the CH3 . to form CH3Cl.
Elimination Reactions
An elimination reaction typically involves the removal of two substituents a substrate in either a one or two-step mechanism. The one-step mechanism is the E2 reaction, and the two-step mechanism is the E1 reaction. The numbering scheme relates to the kinetics of the reaction, i.e., bimolecular and unimolecular, respectively.
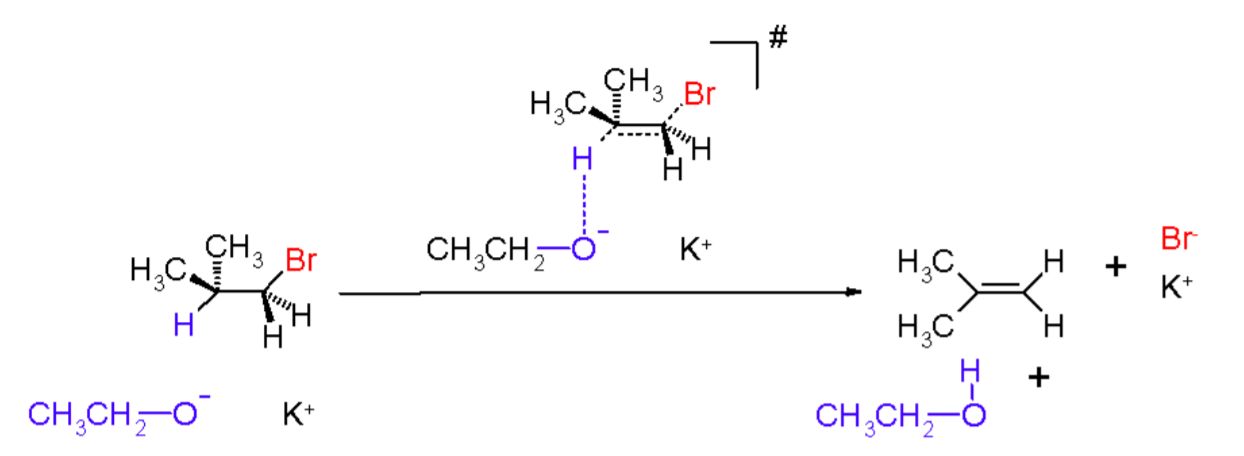
Elimination reactions are important because they are among a very few set of useful reactions that generate unsaturation within organic molecules. They are the opposite of addition reactions and involve homolytic or heterolytic dissociation of molecular groups on adjacent (vicinal) atoms leading to increased bond order on these vicinal atoms.
For example,RCH2CH2OH → R-CH=CH2 +H2O.This latter reaction requires the expulsion of a water to lead to one degree of unsaturation. The presence of a leaving group is a pre-requisite for these types of reactions.
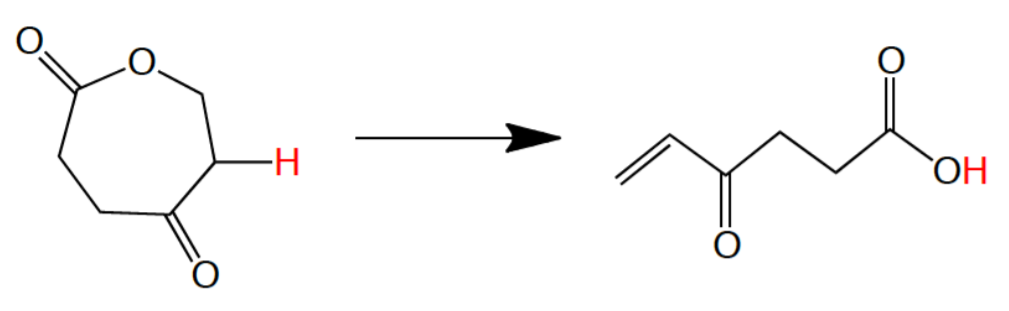
In Figure \(\PageIndex{8}\), the opportunity to retain a hydrogen on the final structure is an unusual mechanistic phenomenon. The conversion relies on a strict orbitally conserved concerted mechanism in which the degree of unsaturation (terminal vinyl group) is generated in tandem with the abstraction of the proton by the lactonic ring oxygen. Such a sigmatropic shift would be over 3 atoms (hence, an unusual 1,3-hydride shift).
Pericyclic Reactions
A pericyclic reaction (Figure \(\PageIndex{9}\) ) is a reaction in which the transition state of the molecule has a cyclic geometry and progresses in a concerted manner. They are typically rearrangement reactions. The major classes of pericyclic reactions are electrocyclic, cycloaddition, sigmatropic, group transfer, cheletropic, and dyotropic, all of which are considered to be equilibrium processes.
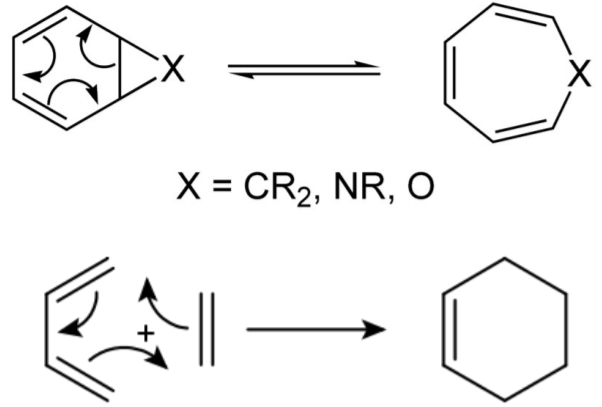
These types of reactions are governed by Frontier Molecular Orbital (FMO) Theory. Frontier molecular orbitals for a molecule are so called because the orbitals are the “frontier” or vanguard (leaders) of electron occupation. These are known classically as the highest-energy occupied (HOMO) and lowest-energy unoccupied (LUMO) molecular orbitals. The HOMO is characterized by nucleophilic or electron donation, whereas the LUMO is the opposite. Chemical reactions and resonance are very successfully described by the overlap between a filled HOMO and an empty LUMO.
Acidity and basicity are terms used in FMO Theory very broadly. Acidity refers to a ligand, metal center, or orbital ability to accept electron density (from electron sources that include Brønsted bases). Basicity refers to a ligand, metal center, or orbital ability to donate electrons (to electron sinks that include Brønsted acids). The theory distinguishes among σ-acids, σ-bases, π-acids, and π-bases. The first two, σ-X, accept or donate electrons in a σ manner, aligned head on with another orbital, whereas the latter two, π-X, accept or donate electrons in a π manner, aligned side by side with another orbital.
A pericyclic reaction typically is unimolecular and zero order with respect to kinetics. As shown below, the bicyclic on the left is highly strained because of the 1,2 bridging and undergoes an electrocyclic RAR to relieve the steric stress associated with the heterocyclic cyclopropanoid moiety.
Diels-Alder
This is a [4+2] cycloaddition as already mentioned that occurs between a conjugated diene and a substituted alkene known as a dienophile to form a substituted cyclohexene. It was first described by Otto Diels and Kurt Adler in 1928 that lead to the Nobel Prize in Chemistry in 1950. It is a reliable method for forming 6- membered systems with good regio- and stereochemical control. The concept has been applied to other π-systems to provide the corresponding heterocycles (hetero-Diels–Alder reaction).
1,3-Dipolar Cycloaddition
This reaction occurs between a 1,3-dipole and a dipolarophile to form a five-membered ring (Figure \(\PageIndex{10}\) ). The reaction is sometimes referred to as the Huisgen cycloaddition which specifically describes the 1,3-dipolar cycloaddition between an organic azide and an alkyne to generate 1,2,3-triazole. Currently, this reaction is an important route to the region- and stereospecific synthesis of fivemembered heterocycles.
Cope Rearrangement (RAR)
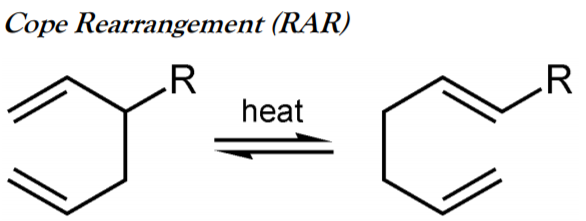
The Cope RAR is an extensively studied [3,3]-sigmatropic RAR of 1,5-dienes developed by Arthur Cope. For example, 3-methyl-1,5-hexadiene heated to 300 °C yields 1,5- heptadiene (see Figure \(\PageIndex{11}\) ).
Oxidation/Reduction Reactions
The term “redox”, short for reduction–oxidation reaction, is used to describe the complete set of chemical reactions occurring for electron transfers leading to changes in oxidation states. Redox reactions require a reduction and complementary oxidation processes. The chemical species (reductant) where the electron is removed is oxidized, while the counter species (oxidant) to where it is added is reduced:
- Oxidation is the loss of electrons or increase in oxidation state;
- Reduction is the gain of electrons or a decrease in oxidation state.
As an example, in an electrolytic cell (shown in Figure \(\PageIndex{12}\) ), a complete redox (cycle) is shown.
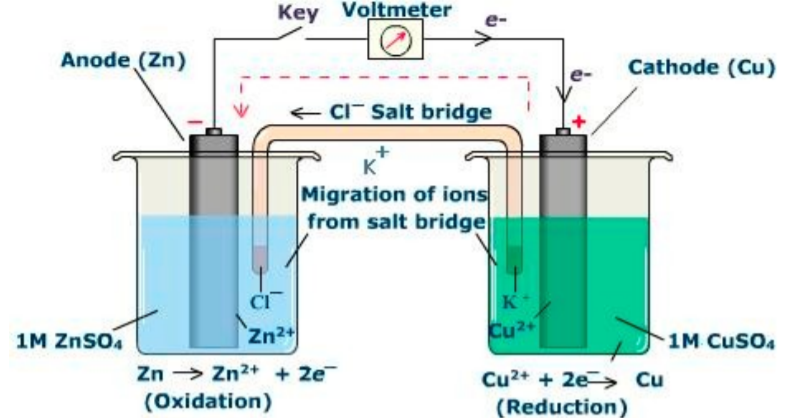
In the example, zinc is being oxidized from Zn0 → Zn+2 which is a net loss of two electrons per atom. The oxidized zinc solubilizes in the anodic solution and proceeds to pull over an anion from the cathodic solution through the electromotive force of the two electrons that it lost. In other words, the zinc cation is balanced by the anions (likely two chloride anions) in the cathodic solution. Simultaneously, copper sulfate cations (cupric cations) are reduced by the net influx of the two electrons. A redox reaction can occur relatively slowly, for example, in the case of rust, or more rapidly, as in fires.