
3. Qualitative and Quantitative Analysis
- Page ID
- 74167

Qualitative Analysis
Issues to consider
Question: What is the GOAL of the analysis and WHAT ELEMENTS do we want to look for (toxic elements such as As, Cd, Hg, Pb; nutrient elements such as Ca, Fe)?
Answer: Define the problem (what to measure, typical concentration range, required detection limit, accuracy, precision, etc.)
Question: Are there any potential SPECTRAL OVERLAPS with other elements in sample?
Answer: Compare line energies of target elements and other elements to identify any possible interferences
Question: If we get a “positive” (detection of a toxic element), do we know for certain that it is IN THE SAMPLE and not in the product packaging or the background materials used to hold the sample?
Answer: Measure what you want to measure and be sure to do “blanks”
Question: How do we know that the analyzer software is not giving ERRONEOUS RESULTS (false positives or false negatives)?
Answer: Users must evaluate the spectrum to verify the reported results – positive identification of an element requires observation of two peaks at energies close to their tabulated values
Spectra for positive, tentative, and negative identifications
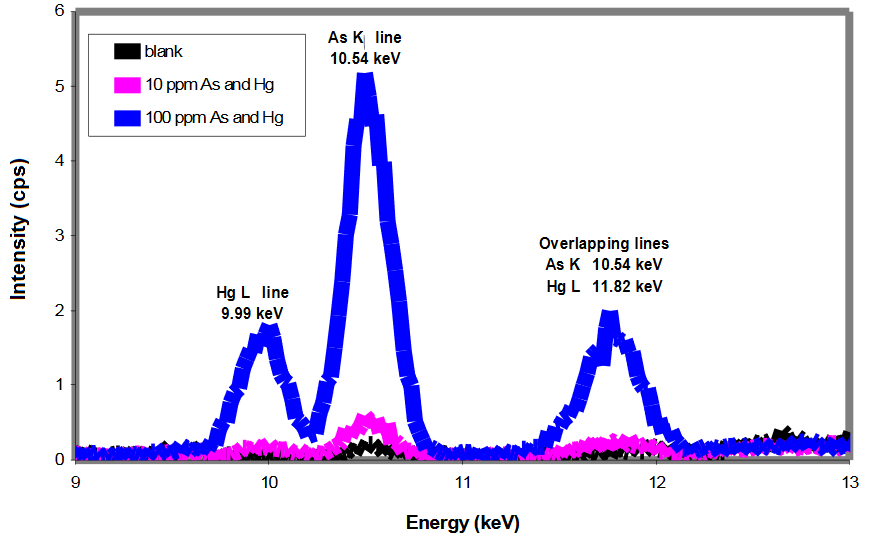
- As and Hg clearly present in blue spectrum (see both α and β peaks)
- As and Hg possibly present in purple spectrum (β peaks barely > blank)
- As and Hg not present in black spectrum (no visible peaks)
False positive for Pb in baby food cap
Spectrum
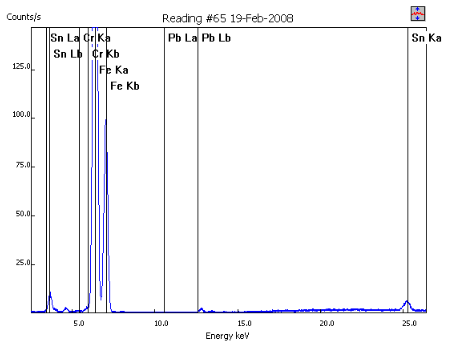
Closeup of Pb lines
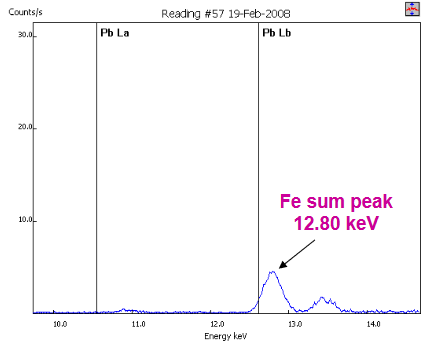
- User acquired sample spectrum near lid (>10% Fe), which gave Fe sum peak at 6.40 keV * 2 photons = 12.80 keV
- Vendor algorithm incorrectly identified Pb in this sample at over 2000 ppm (detection and quantitation based on signal at the Pb Lβ line at 12.61 keV, zero intensity of Pb Lα line at 10.55 keV not considered by algorithm)
- Be wary of analyzer software and be sure to avoid potential false positives such as this by evaluating the spectrum to confirm the presence of an element
False negative for U in tableware

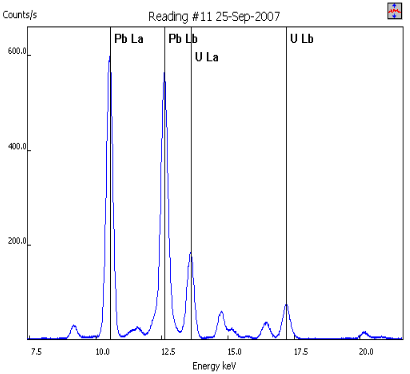
- Vendor algorithm did not identify U in this sample (algorithm not intended to attempt this identification of this and other relatively uncommon elements)
- Lack of manual interpretation of the spectrum of a product containing only U would have led to the assumption that it was safe
- Be wary of analyzer software and be sure to avoid potential false negatives such as this by evaluating the spectrum to identify unexplained peaks
Conclusions on Qualitative Analysis
Vendor software on commercial XRF analyzers are usually reliable in identifying which elements are present in a sample, but are not foolproof and an occasional false positive or false negative is possible
FALSE POSITIVES (element detected when not present)
- Due to limitations in the vendor software, which make not take into account line overlaps, sum peaks, escape peaks
- Users must confirm positive detection of an element based on the observation of two peaks centered within ±0.05 keV of the tabulated line energies for that element at the proper intensity ratio (5:1 for K lines, 1:1 for L lines)
FALSE NEGATIVES (element not detected when present)
- Due to limitations in the analyzer software, which may not be set up to detect all possible elements in the periodic table
- Unlikely occurrence for toxic elements such as As, Hg, Pb, and Se, more common for rare elements such as U, Th, and Os
- Users must identify “non-detected” elements through manual interpretation of the spectrum
Quantitative Analysis
Issues to consider
Question: Are the element CONCENTRATIONS within the detection range of XRF (% to ppm levels)?
Answer: Define the problem, research sample composition, or take a measurement
Question: What sort of SAMPLE PREP is required (can samples be analyzed as is or do they need to be ground up)?
Answer: Consider sample - is it homogeneous?
Question: For SCREENING PRODUCTS, are semi-quantitative results good enough? For example, if percent levels of a toxic element are found in a supplement, is this sufficient evidence to detain it or to initiate a regulatory action?
Question: For ACCURATE QUANTITATIVE ANALYSES, what is the most appropriate calibration model to use for the samples of interest (Compton Normalization, Fundamental Parameters, empirical calibration, standard additions)?
Types of Calibration Models
VISUAL OBSERVATION (rough approximation, depends on many variables)
- Peak intensity >100 cps corresponds to concentrations >10,000 ppm (% levels)
- Peak intensity of 10-100 cps corresponds to concentrations of ~100-1000 ppm
- Peak intensity of 1-10 cps corresponds to concentrations ~10-100 ppm
- Peak intensity < 1 cps corresponds to concentrations ~1-10 ppm
FUNDAMENTAL PARAMETERS (aka FP or alloy mode)
- Uses iterative approach to select element concentrations so that modeled spectrum best matches samples spectrum (using attenuation coefficients, absorption/enhancement effects, and other known information)
- Best for samples containing elements that can be detected by XRF (i.e., alloys, well characterized samples, and samples containing relatively high concentrations of elements)
COMPTON NORMALIZATION (aka CN or soil mode)
- Uses “factory” calibration based on pure elements (i.e., Fe, As2O3) and ratioing the intensity of the peak for the element of interest to the source backscatter peak to account for differences in sample matrices, orientation, etc.
- Best for samples that are relatively low density (i.e., consumer products, supplements) and samples containing relatively low concentrations of elements (i.e., soil)
OTHER MODES – thin film/filters, RoHS/WEEE, pass/fail, etc.
- Beyond scope of these training materials
EMPIRICAL CALIBRATION
- Involves preparation of authentic standards of the element of interest in a matrix that closely approximates that of the samples
- Provides more accurate results than factory calibration and Compton Normalization
- Note that the XRF analyzer can be configured and used with this type of calibration to give more accurate results for the elements and matrices of interest
- Usually reserved for laboratory analyses by trained analysts, using a high purity metal salt containing the element of interest, an appropriate matrix, homogenization via mixing or grinding
STANDARD ADDITIONS
- Involves adding known amounts of element of interest into the sample
- Provides most accurate results as the standards are prepared in the sample matrix as the sample
- Usually reserved for laboratory analyses by trained analysts, and even then used only as needed as this is labor intensive and time consuming
Effect of Concentration
Spectra of As standards in cellulose
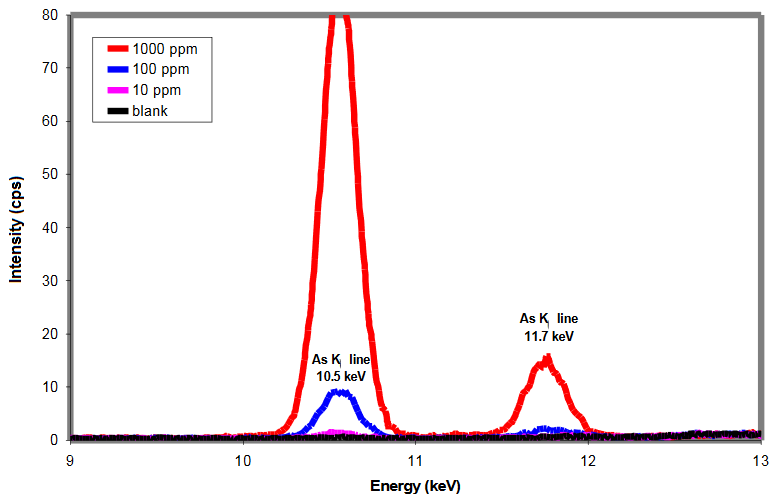
- Intensity is proportional to concentration
- Detection limits depend on element, matrix, measurement time, etc.
- Typical detection limits are as low as 1 part per million (ppm)
Peak Intensity vs Concentration
Linearity falls off at high concentrations
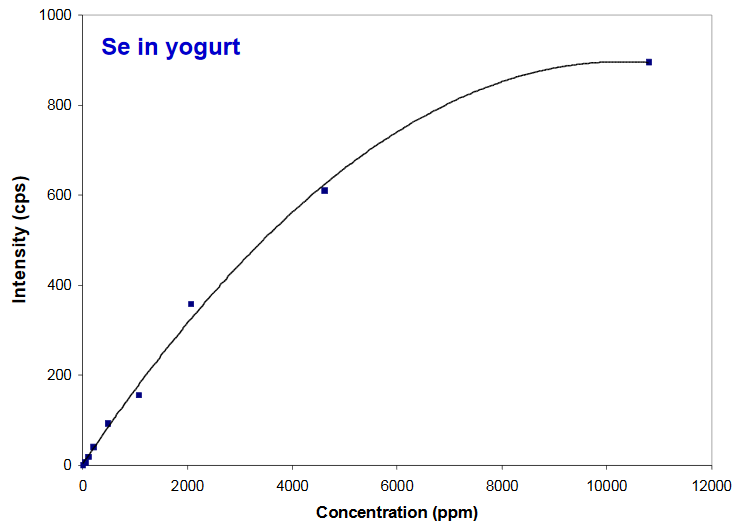
- Response becomes nonlinear between 1000-10,000 ppm
- Use of Compton Normalization will partially correct for this
Compton Normalized Intensity vs Conc.
Linearity improves through use of “internal standard”
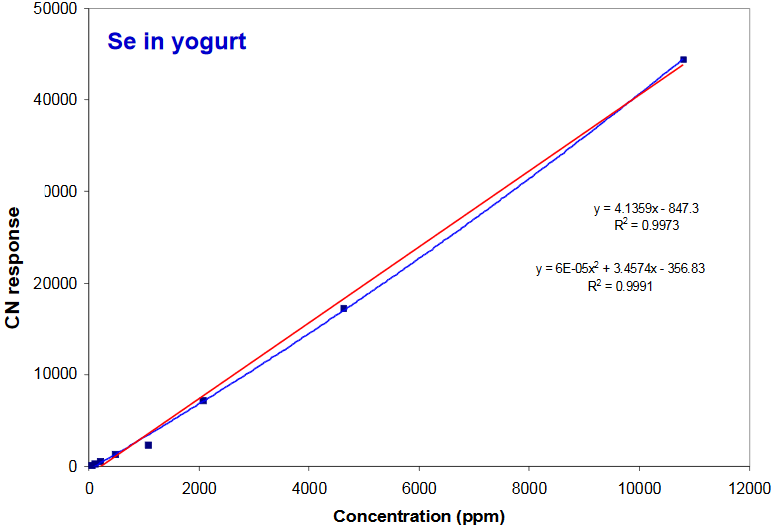
- Use of Compton Normalization (X-ray tube source backscatter from sample) partially corrects for self absorption and varying sample density
Quantitative Analysis at High Conc’s
Cr standards in stainless steel for medical instrument analysis
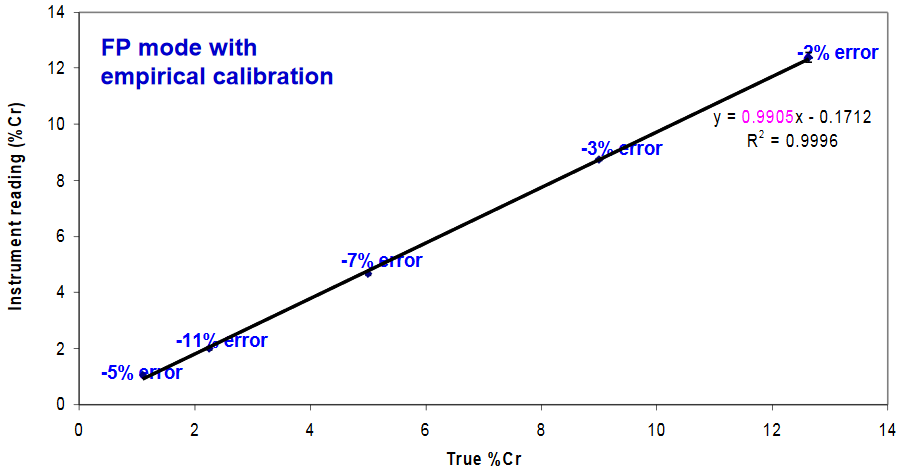
- Although Fundamental Parameters based quantitation gives fairly accurate results, it also gives determinate error (consistently negative errors)
- Determination of Cr in surgical grade stainless steel samples using an XRF analyzer calibrated with these standards gave results that were statistically equivalent to flame atomic absorption spectrophotometry
- For determining % levels of an element, use Fundamental Parameters mode
Quantitative Analysis at Low Conc’s
As, Hg, Pb, and Se standards in cellulose for supplement analysis
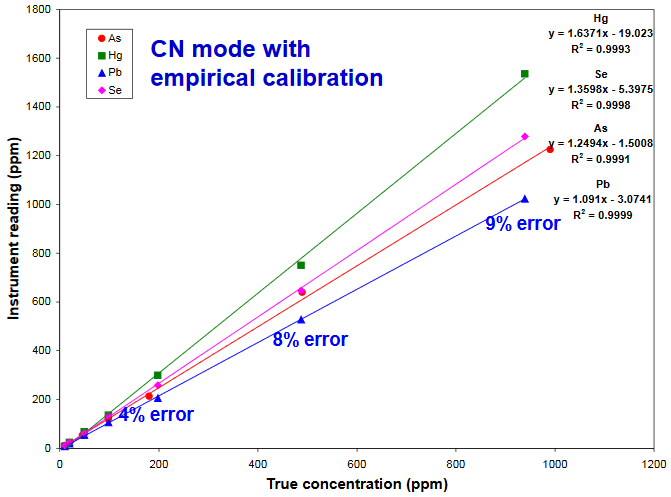
- Although Compton Normalization based quantitation gives fairly accurate results, it can also give significant determinate error (slopes > 1)
- Determination of Pb in supplements using an XRF analyzer calibrated with these standards gave results that were statistically equivalent to ICP-MS
- For determining ppm levels of an element, use Compton Norm. mode
Standard Additions Method
Determination of As in grapeseed sample
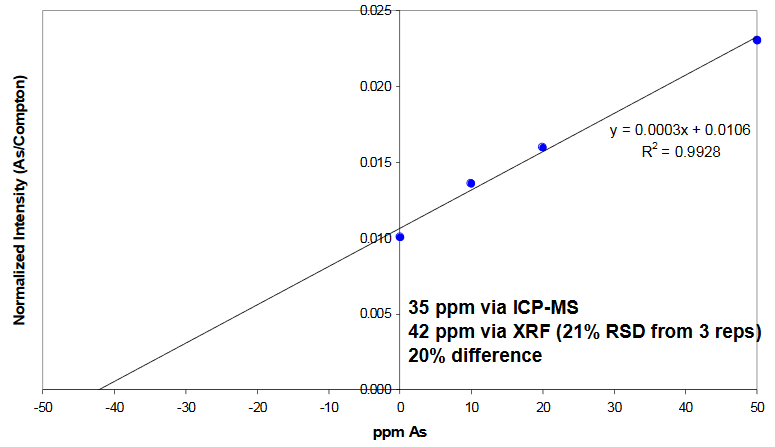
- Typically gives more reliable quantitative results as this method involves matrix matching (the sample is “converted” into standards by adding known amounts of the element of interest)
- This process is more time consuming (requires analysis of sample “as is” plus two or more samples to which known amounts of the element of interest have been added)
Effect of Measurement Time
Longer analysis times give better precision and lower LODs
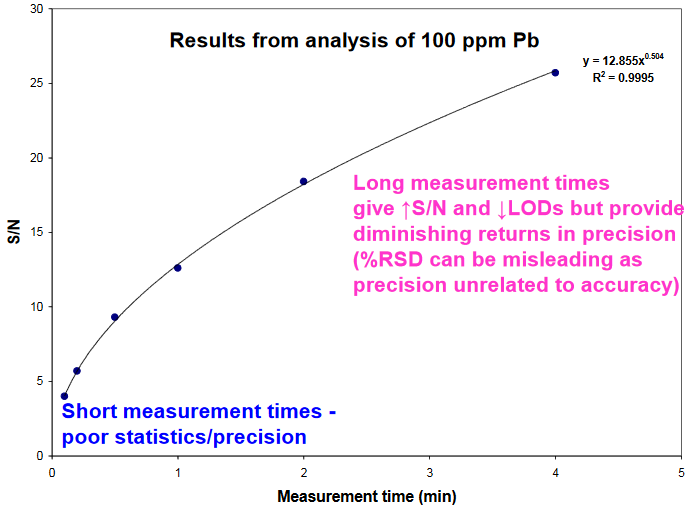
- S/N = mean signal / standard deviation of instrument response (noise)
- As per theory, S/N is proportional to square root of measurement time
- 1-2 min measurement gives a good compromise between speed and precision
- Longer measurement times give better S/N and lower LODs
Conclusions on Quantitative Analysis
For field applications, the sample is often analyzed “as is” and some accuracy is sacrificed in the interest of shorter analysis times and higher sample throughput, as the more important issue here is sample triage (identifying potential samples of interest for more detailed lab analysis)
- Use FP mode to analyze samples that contain % levels of elements
- Use CN mode to analyze samples that contain ppm levels of elements and have varying densities
For lab applications, more accurate quantitative results are obtained by an empirical calibration process
- Grind/homogenize product to ensure a representative sample
- Calibrate the analyzer using standards and/or SRMs
- Use a calibration curve to compute concentrations in samples
- When suitable standards are not available or cannot be readily prepared, consider using the method of standard additions
For either mode of operation, getting an accurate number involves much more work than implied in the “point and shoot” marketing hype of some XRF manufacturers