
Section 3C. Peptide Mass Mapping for Protein Identification
- Page ID
- 80415

Peptide mass mapping is a technique that uses powerful search engines (e.g. Mascot) to identify a protein from mass spectrometry data and primary sequence databases. The general approach is to take a small sample of the protein and digest it with a proteolytic enzyme, such as trypsin. Trypsin cleaves the protein after lysine and arginine residues. The resulting mixture of peptides is analyzed by MALDI-TOF mass spectrometry.
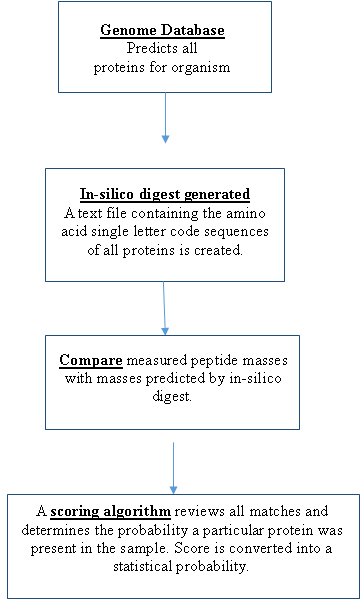
The experimental mass values of the peptides are then compared with theoretical peptide mass values. Theoretical mass values of peptides are obtained by using the genome sequence for an organism and predicting all the proteins that can be expressed. Once all the proteins are predicted then the cleavage rules for the digest enzyme are applied and the masses of the resulting peptides calculated by the computer (in-silico digest). By using an appropriate scoring algorithm, the closest match or matches can be identified. If the "unknown" protein is present in the sequence database, then the aim is to pull out that precise entry. If the sequence database does not contain the “unknown” protein, then the aim is to pull out those entries which exhibit the closest homology, often equivalent proteins from related species. The steps in peptide mass mapping are outlined in the flow chart.
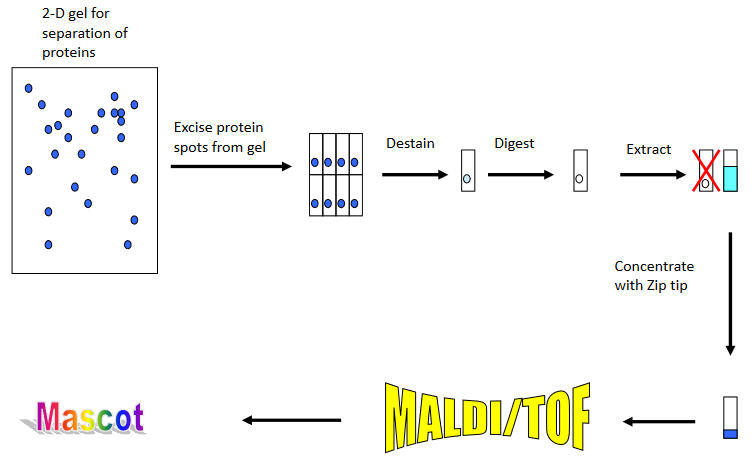
The analysis of a complex mixture of proteins from an organism always involves some type of separation step to isolate a certain protein. The separation methods frequently used are two dimensional (2D) gel electrophoresis or liquid chromatography. The figure shows the experimental workflow used to identify a protein spot from a 2D gel.
1. In your own words, describe the general principle of peptide mass mapping for protein identification.
A. The sequence of DNA bases in an organism’s genome is used to predict all the proteins the organism will synthesize. Once the proteins are predicted, then the cleavage rules for a specific enzyme such as trypsin are applied and the masses of the different peptides are predicted for each protein. Experimentally, a protein is removed from a gel and digested with an enzyme. The masses of the peptides from the digest are measured using MALDI-TOF mass spectrometry and compared with the theoretical peptide matches. A statistically significant number of peptide mass matches will identify a protein.
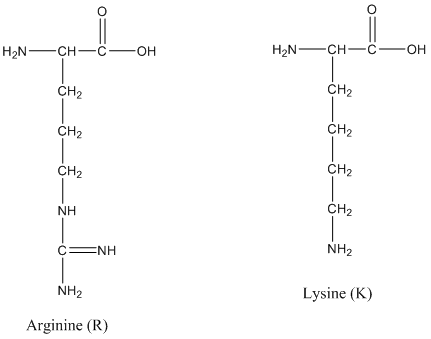
The enzyme trypsin is frequently used to digest proteins in the peptide mass mapping technique. Trypsin cleaves the amide bond after lysine (K) and arginine (R) residues. K and R make up about 10% of the amino acids in a protein and digesting with trypsin typically results in peptides in a useful range mass range for mass spectrometry (500-3,000 Da). (Remember: The mass unit amu is the same as Da.)
The structures of lysine and arginine are shown.
1. What is the purpose of digesting the protein with trypsin? (Hint: Think about how mass spectrometry is used to identify and/or elucidate the structure of small organic molecules such as caffeine).
A. The molecular weight of a protein is not a unique characteristic. There are many proteins that could have similar or identical molecular weights and the molecular weight alone cannot be used for identification. Digesting the protein with trypsin cleaves the protein into smaller fragments in a predictable way. The matching of many masses (from different peptide fragments) will be a unique characteristic that can be used to identify a protein. Peptide mass mapping is often referred to as a “peptide fingerprint” because just like a fingerprint analysis there must be several unique similarities for a match to be made.
2. Why is a well annotated genome for the organism of interest needed in the peptide mass mapping technique for identifying a protein?
A. A protein is identified by taking experimentally measured peptide masses and matching them with predicted masses. The computer predicts the peptide masses in the following way. First, the sequence of DNA bases is used to predict all the proteins produced by a given organism. Then, peptide masses for each protein are predicted by cleaving the protein after lysine and arginine.
There would not be theoretical masses to match to experimental data without the DNA sequence for the organism.
3. Classify the side chains of R and K as acidic or basic.
A. Basic
4. Digesting the protein with trypsin ensures that there is an R or K residue in each peptide. Why is this helpful for MALDI-TOF analysis? (Hint: Think about the function of the matrix in MALDI).
A. The basic residues readily accept a proton from the matrix giving the peptide a positive charge. The positive charge is needed so the ion can be accelerated with a voltage in the TOF mass analyzer.
5. a. Can the following two peptides with the same amino acid composition be distinguished using MALDI-TOF mass spectrometry?
Peptide 1: GASPVRTCILKMHFY
Peptide 2: GMFHRATIKYPVCSL
A. No they cannot be distinguished because they have the same mass.
b. Calculate the expected (monoisotopic) masses if the enzyme trypsin was used to digest peptides 1 and 2. A table of amino acid masses is provided. Can the peptides be distinguished after digestion with trypsin?
(Refer to the Introduction section of this module if you need assistance in calculating the mass of a peptide or defining the difference between a monoisotopic mass and average mass.)
A.
Peptide 1 has the following masses after digestion with trypsin.
GASPVR (585.33 amu)
TCILK (146.11 amu)
MHFY (596.25 amu)
Peptide 2 has the following masses after digestion with trypsin.
GMFHR (646.31 amu)
ATIK (431.28 amu)
YPVCSL (593.29 amu)
Yes, peptide 1 and 2 can be distinguished once they are digested with trypsin. The masses of the three smaller peptides are different.
6. There are other enzymes that could be used to digest the proteins.
Pepsin is most efficient in cleaving peptide bonds between hydrophobic amino acids (leucine) and aromatic amino acids such as phenylalanine, tryptophan, and tyrosine. Pepsin is less specific and results in many small peptides. Would pepsin be a good choice for digesting proteins for peptide mass mapping? Explain your reasoning.
There are also enzymes that are highly specific and result in only a few cleavage sites in a protein. Is a highly specific enzyme that creates a few large peptides be a good choice for peptide mass mapping? Explain your reasoning.
A. An enzyme that results in many small peptides would be not be ideal for peptide mass mapping because there would be a significant chance that other proteins would have small peptides with the exact same amino acid composition. The goal is to produce peptides that are unique to one protein.
An enzyme that only has a few cleavage sites and results in a few large peptides is not ideal for peptide mass mapping either. Multiple peptide matches are required for identification. The greater number of matches the higher the probability that the protein has been identified correctly. A few large peptides would only give a few masses to be matched
Table 2. Molecular weight information for all twenty naturally occurring amino acids.
|
Amino Acid |
Single-Letter Code |
Residue MW (amu) |
Amino Acid MW (amu) |
|---|---|---|---|
|
glycine |
G |
57.02 |
75.03 |
|
alanine |
A |
71.04 |
89.05 |
|
Serine |
S |
87.03 |
105.04 |
|
proline |
P |
97.05 |
115.06 |
|
Valine |
V |
99.07 |
117.08 |
|
threonine |
T |
101.05 |
119.06 |
|
cysteine |
C |
103.01 |
121.02 |
|
isoleucine |
I |
113.08 |
131.09 |
|
leucine |
L |
113.08 |
131.09 |
|
asparagine |
N |
114.04 |
132.05 |
|
aspartic acid |
D |
115.03 |
133.04 |
|
glutamine |
Q |
128.06 |
146.07 |
|
Lysine |
K |
128.09 |
146.11 |
|
glutamic acid |
E |
129.04 |
147.05 |
|
methionine |
M |
131.04 |
149.05 |
|
histidine |
H |
137.06 |
155.07 |
|
phenylalanine |
F |
147.07 |
165.08 |
|
arginine |
R |
156.10 |
174.11 |
|
tyrosine |
Y |
163.06 |
181.07 |
|
tryptophan |
W |
186.08 |
204.09 |