
Metals Analysis by Anodic Stripping Voltammetry
- Page ID
- 142892

After reading about ASV and working through this learning module, you should be able to:
- Describe some advantages of anodic stripping voltammetry.
- Outline the general steps in an ASV experiment.
- List the important parameters that affect sensitivity and detection limits in an ASV experiment.
- Perform calculations for determination of metal ion content in unknowns analyzed by ASV. These calculations will utilize the single standard addition quantitation method.
Let’s say that you wanted to determine the heavy metal identities and concentrations in sediment and suspended solid samples obtained from present day Lake Nakuru. What technique would you use for this determination? You could use Atomic Absorption Spectroscopy (AAS) as was done in the article (Nelson 1998), but another technique, which is particularly good at detecting metals at the ppb level is anodic stripping voltammetry (ASV). This learning module will focus on ASV.
Purpose
The purpose of this learning module is to introduce students to the electrochemical technique of anodic stripping voltammetry (ASV). The content is appropriate for students in a course in analytical chemistry or instrumental analysis. This introduction will be achieved via an inquiry-based learning style.
Content specific to voltammetric methods and ASV can be found in the citations listed in the reference section of this document and are available free of charge at the following websites:
- in the Analytical Electrochemistry module (Kelly 2009) at the ASDL website (click here to link directly to the ASV section)
- A chapter on voltammetric techniques can be found in the eText Analytical Chemistry 2.0 found in the ASDL collection (Chapter 11; specifically section 11.4 discusses stripping voltammetry).
- Additionally, you can perform a search with the key words anodic stripping voltammetry at the ASDL website (click here for the voltammetry section of the ASDL collection accessed on June 26, 2012).
Theory
Read the Anodic Stripping Voltammetry section of the Analytical Electrochemistry eLearning module and answer the following questions. If you have no background in voltammetric measurements and instrumentation, read the Basics of Voltammetry section of the Analytical Electrochemistry module. Once you have read the background material, the questions below will help you ascertain and apply important aspects of ASV theory and practice.
Q1. How would you prepare the solid samples obtained from Lake Nakuru for an ASV analysis? In what physical form should they be for ASV analysis? What are the expected concentration levels in your samples from Lake Nakuru?
All voltammetric techniques require a working electrode – this is where the electrochemical reactions occur and where the analytical signal is generated.
Q2. What is the most common type (i.e. material used) of working electrode used in ASV? You should be able to find this information in the material referenced in the Theory section of this document.
An advantage of mercury as an electrode material is that mercury forms amalgams (solid solutions) with many metals allowing preconcentration of more of the analyte metal at the electrode surface. The metal in solution is cationic, but the mercury amalgam forms with the neutral metal analyte.
Q3. What must happen to the metal at the electrode surface to form the neutral species?
ASV is useful for detecting metal ions at the ppb level. The preconcentration step is key for achieving these low detection limits. When this preconcentration step utilizes electrochemical accumulation, the metal must be reduced and form an amalgam in the mercury (drop or film). In a mercury electrode, the concentration of metals is 100-1000 times that in solution.
Q4. Look up the reduction potentials of the following metals of interest (Cu2+, Pb2+ and Zn2+). You can find a table of reduction potentials in the back of most analytical textbooks. These are generally reported with respect to the standard hydrogen electrode (SHE). We will continue this convention throughout the module. You can also find a table of values here (from the online textbook Analytical Chemistry 2.0 Appendix 13, p. 1107). Would you be able to discriminate between these three metals based on their reduction potentials?
Judging from the separation between potentials it appears that these metals could be resolved in an ASV measurement. However, in practice it can be difficult to discriminate between Cu and Zn. Intermetallic compounds between Cu and Zn can form in the Hg amalgam, particularly at thin Hg film electrodes (Copeland 1974). To remove one of the interfering ions during a determination, a complexing agent can be added. For example, during the determination of zinc, sulfide can be added to selectively complex with copper (Lau 1998).
Q5. What potential (versus SHE) would be necessary to simultaneously pre-concentrate zinc and copper?
Q6. Using the symbol M2+ to represent a divalent metal cation, write the appropriate redox reaction for the reduction of the cation at a mercury electrode. In addition to the applied potential, what other experimental variable is used to “drive” the metal cation to the electrode?
The following figure depicts the potential-time waveform for an ASV experiment.
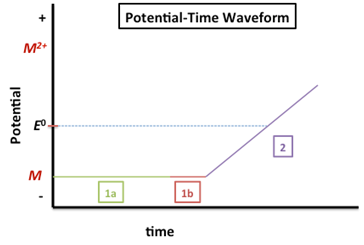
The potential labeled 1a in Figure 1 is the deposition/accumulation/preconcentration potential. A DC potential is applied at 300-500 mV more negative than the E0 of the metal with the most negative reduction potential (if analyzing a mixture). This would be ~-1.1 V since the Zn2+ (E0 = -0.7618 V) has the most negative reduction potential of the three metals listed in Table 1. The reduction reaction, indicating the amalgamation with mercury is shown below.
\[\ce{M^2+(aq) + 2e- \xrightarrow{Hg} M(Hg)} \nonumber\]
The duration of the deposition step is determined by the analyte concentration – it should be longer for lower concentration analytes. Generally, the deposition time is between 1 and 30 minutes. The solution containing the analyte is stirred during this step (or the electrode can be rotated) to ensure faster and more reproducible transport to the electrode.
The interval indicated by 1b is the quiescent time (quiet time). During this period the potential is still applied but the stirring is stopped for 30-60 sec prior to analysis.
In the region labeled 2 in Figure 1, the potential is scanned to oxidize the metal back to its original state. The linear ramp as shown in Figure 1 is the simplest method for re-oxidizing the amalgamated metal:
\[M(Hg) \rightarrow M^{2+}(aq) + 2e^- + Hg \nonumber\]
The resulting plot of current versus potential for this oxidation process is called a voltammogram. The observed peak current is proportional to the analyte concentration.
Q7. In the space below, sketch out the voltammogram (plot of current versus potential) expected from the experiment outlined in Figure 1.
Q8. What values would you choose as the start and end of the potential ramp for an analysis of zinc?
Q9. Summarize the quantitative and qualitative nature of the ASV experiment.
Q10. The Lake Nakuru samples will most likely contain measurable concentrations of copper, lead and zinc. Describe how multiple metals can be analyzed in a single ASV experiment. Draw what the voltammogram would look like. What starting and ending potentials would you use?
ASV is very sensitive to experimental conditions. Important variables include electrode size (described by Hg film area or Hg drop size), deposition time, rest time, stirring rate, and scan rate during stripping. An example set of experimental conditions is given below:
- Working electrode = Hg or Hg film
- Deposition time = 5 min (depends on concentration)
- Deposition potential = -1.1 V (This chosen potential is with respect to the standard hydrogen electrode. The Potentiometry e-learning module found in the ASDL collection provides a review of Reference Electrodes. The Electrochemical Methods Chapter in Analytical Chemistry 2.0 also discusses various reference electrodes.)
- Rest time = 30 s
- Stirring rate = 600 rpm
- Scan rate during stripping = 200 mV/s.
- Final potential (after anodic scan) = 600 mV
Q11. How could the experimental conditions be modified to increase the analyte signal during an analysis? Hint: think about what variables will increase the amount of metal deposited and look at the equation for ip. (This equation can be found in the ASV section of the Analytical Electrochemistry module).
The use of potential waveforms other than the linear scan discussed here can improve detection limits. These are called “step” methods. For example, in the paper Locatelli 1999, differential pulse anodic stripping voltammetry (DPASV) is used for the determination of Cu, Pb, Cd, and Zn. Differential pulse voltammetry improves detection limits by minimizing the background current portion of the current and therefore increasing signal to noise. DPASV is discussed in more detail in the ASV section of the Analytical Electrochemistry module.
Advantages of ASV
Anodic stripping voltammetry, along with certain other electrochemical techniques, is capable of determining the oxidation state of a metal (this is called speciation). For example, as seen in a table of Standard Redox Potentials, copper(I) reduces at 0.520 V and copper(II) reduces at +0.159 V. An ASV experiment could be designed to discriminate between these two oxidation states during the potential stripping scan. As we have observed, ASV is also capable of simultaneous determination of multiple metals. An atomic spectroscopic (AS) technique, such as atomic absorption spectroscopy, could not discriminate between the two oxidation states as all metal in the sample is reduced to neutral gas phase atoms during the atomization step. Additionally, electrochemical techniques generally cost less than their spectroscopic counterparts, and are amenable to miniaturization and to in situ monitoring. ASV requires minimal sample pre-treatment and is generally not affected by high salt content. However, ASV is limited in the number of metals it is able to determine when compared to AS techniques. For example, when Hg-based working electrodes are used, the metal must form an amalgam with mercury. Table 11.11 in Analytical Chemistry 2.0 supplies a list of metals that can typically be determined by ASV.
Special Instructions for trace analysis.
Because of the low concentrations of the metals in samples from many natural systems, special care must be taken during sampling and sample handling. Analytes can be lost via adsorption to sample containers and glassware. Additionally, these containers and glassware can be sources of contamination. Glassware and containers must be thoroughly washed and even soaked in deionized water to remove any possible contaminants. Any solution containing trace metal amounts should be stored in a special Teflon bottle. As previously discussed, electrochemical techniques can provide information about the oxidation state of a metal. However, physical and chemical changes made to the sample can change the original oxidation state. Do not add substances known to be oxidizing or reducing agents to your samples. Check for trace metal impurities in the reagents used.
Quantitation methods. Either the standard addition or external calibration methods can be used for quantitation in ASV. In this module the standard addition method has been used to decrease matrix effects from the lake water. For a review of the standard addition method, see Chapter 5: Standardizing Analytical Methods in Analytical Chemistry 2.0.
Data and Analysis
In the following section, you will review the data reported in Nelson 1998 for the analysis by atomic spectroscopy and XRF of Cr, Cu, Pb, and Zn in suspended solids and dried sediments from Lake Nakuru. Next, you will be supplied with simulated ASV data from Lake Nakuru sediments so that you can compare current levels of the four target metals to those obtained in the 1996 study. As a starting point, let us consider Figures of Merit common to the ASV experiment, as well as some practical considerations for application of the technique.
Figures of Merit, Anodic Stripping Voltammetry
Precision and Accuracy. You can typically expect a precision of ~2-4% relative standard deviation and similar accuracy in the range of 2-5% relative error.
Detection Limits and Linearity. You have seen that ASV is an extremely sensitive technique, with detection limits commonly in the ppb range. Part per trillion detection limits can be reached for some analytes by using an increased deposition time. ASV is typically performed on dilute solutions and the linearity is generally over two orders of magnitude.
Interferences. The metals being analyzed must have redox potentials sufficiently different so that the peaks are resolved and proper quantitation can be performed. Typically up to five metals can be simultaneously determined by ASV. Only certain metals can be determined by ASV. Common metals are Cd, Pb, Zn, Tl, In, and Cu.
Practical Considerations for Anodic Stripping Voltammetry
Cost. ASV is a relatively inexpensive technique. The electrochemical instrumentation and electrodes are a fraction of the cost of other metal analysis methods (e.g. ICP and AA systems).
Speed. The per sample run time depends on the detection limits required. The lower the sample concentrations, the longer the analysis time. The primary factor is the deposition time. For ppm to ppb concentrations this can be a few seconds to a minute. For ppb-ppt detection limits, the deposition time can be a few to many minutes.
Data from Nelson Study, 1998
Table 2 gives the levels for chromium, copper, lead, and zinc found in Lake Nakuru sediments and suspended solids collected by Nelson, et al. in 1996. (Nelson 1998)
Table 2. Metal Concentrations in Lake Nakuru Sediments and Suspended Solids (Nelson 1998)
| Concentration in dry sediments (µg/g) | Concentration in suspended solids (µg/g) | |||
|---|---|---|---|---|
| Trace metal | Average (11 sites) | Range | Average | Standard deviation |
| Chromium | 67 | 10 – 280 | 8.3 | 0.82 |
| Copper | 24 | 5 – 95 | 19 | 2.9 |
| Lead | 22 | 4 – 100 | 11.7 | 0.9 |
| Zinc | 147 | 44 – 630 | 74 | 31 |
Q12. Are the levels observed for each of these four metals in sediments and suspended solids amenable to analysis by ASV? Note that the Nelson paper does not prepare samples for ASV analysis. In your answer, consider the sample preparation steps described by Locatelli 1999, which is described in the next section.
Sample Preparation and Data Analysis.
In this section, you will be given the chance to obtain practical experience evaluating ASV data representative of the current levels of the metals of interest in simulated Lake Nakuru sediment and suspended solid samples. These samples were collected and prepared in a manner similar to that described in the literature (Locatelli 1999). Briefly,
“Two samplings . . . were made with a plexiglass device. Single carrots of superficial sediment (height 5 cm) were drawn out and stored in polyethylene bottles. The samples, dried at 60 C for 48 hr, were sieved through a 40 mesh sieve to eliminate coarse material, then through a second 150 mesh sieve, and lastly powdered by means of a corundum ball mill. The sediments were then mineralized with the acidic mixture HNO3-HCl. . . .
Approximately 1.0 g, weighed accurately in a pyrex digestion tube, was dissolved in 10 mL of 1:1 diluted 69% (m/m) nitric acid. The tube was inserted into the cold home-made block digester, raising gradually the temperature up to 95 C and keeping it for all the time of mineralization. After 15 min 10 mL, and after 30 min further addition of 5 mL of concentrated nitric acid were made. At the end, 5 mL of 37% (m/m) hydrochloric acid was added, maintaining the temperature at 95 C for a further 15 min. After cooling the digest was diluted to 100 mL. The solutions so obtained were diluted 1:20 before voltammetric and spectroscopic measurements.
Voltammetric curves were recorded with a Metrohm Model VA 646 polarographic processor equipped with a Metrohm Model VA 647 Stand. An Ag/AgCl (KCl sat.) electrode and a platinum wire were used as reference and auxiliary electrode, respectively. The voltammetric cell was rinsed every day with suprapure concentrated HNO3 to avoid metal contamination of the sample. The solutions were thermostated at 20±0.5 C and deaerated with pure nitrogen prior to analysis, while a nitrogen blanket was maintained above the solutions during analyses. The solutions were stirred with a Teflon-coated magnetic stirring bar in the electrolysis step.”
Differential Pulse ASV measurements. The instrumental experimental conditions used are shown in the table below.
Table 3: Experimental conditions for the determination of copper, lead, and zinc by DPASV in sediments*
| Ed | Electrolysis potential (mV/Ag, AgCl, KCl satd.) | –1150 |
| Ef | Final potential (mV/Ag, AgCl, KCl satd.) | +50 |
| dE/dt | Potential scan rate (mV/s) | 10 |
| f | Pulse repetition (s) | 0.1 |
| ΔE | Pulse amplitude (mV) | 50 |
| v | Pulse duration (ms) | 40 |
| ts | Sampling time (ms) | 8 |
| td | Electrolysis time (s) | 240 |
| tr | Rest time (s) | 15 |
| t | Purging time prior of the electrolysis (s) | 300 |
| u | Stirrer speed (r.p.m.) | 600 |
*Note: conditions modified from Locatelli 1999 who were determining Cu, Pb, Cd, and Zn in sediment samples.
Below are simulated sample data acquired for samples taken from Lake Nakuru. Sample preparation was similar as described above by Locatelli 1999. Briefly, 1 g sediment samples were acid digested and diluted to 100 mL. This solution was diluted by a factor of 20 with appropriate electrolyte. The analysis was performed on this second solution. The single standard addition method of quantitation was used (section 5.3.3 in Analytical Chemistry 2.0). A 10.00 mL aliquot of sample solution was pipetted into the sample container and purged with nitrogen as described above. The sample underwent the anodic stripping procedure outlined above. Upon collection of this voltammogram, 100.0 µL of the standard solution (5.000 ppm) was added to the 10.00 mL sample and the ASV procedure was performed on this “spiked” sample. The data for the peak current was recorded in tabular format and is shown below. You will use this data to calculate the concentration of each heavy metal in the sediment samples in µg/g (as was reported in Nelson 1998). Don’t forget to account for any dilutions performed during the sample preparation. You may hand-calculate the concentration for sample 1 then set up a spreadsheet to calculate the concentrations of the metals for all three samples.
Q13. Recently, sediment samples were collected from Lake Nakuru and analyzed by ASV in the manner described above. Given the sample data in Table 4, calculate the current concentration levels of the heavy metals (Cu, Pb and Zn) in the sediment samples obtained from Lake Nakuru. What do the data tell you about the heavy metal concentrations in the sediments at Lake Nakuru? Have the concentrations increased or decreased since the 1996 sampling?
Table 4: Sample Data for Copper, Lead and Zinc Determination in Sediment Samples in Lake Nakuru
|
Sample number |
ip (µA) |
|||||
|
Sample |
Sample + 100 µL spike |
|||||
|
Cu |
Pb |
Zn |
Cu |
Pb |
Zn |
|
|
1 |
0.0985 |
0.1011 |
0.2134 |
0.2000 |
0.3045 |
0.3152 |
|
2 |
0.0996 |
0.1008 |
0.2159 |
0.2015 |
0.3026 |
0.3165 |
|
3 |
0.1000 |
0.1205 |
0.2200 |
0.2099 |
0.3225 |
0.3251 |
Note: All samples analyzed were a 10.00 mL aliquot. The 100.0 µL standard spike contained a target concentration of 5.000 ppm of each metal of interest. The exact concentration (depending on standard mass weighed out) can be determined via calculation.
Data Analysis – Suspended Solids
Q14. Recently, suspended solid samples were collected from Lake Nakuru and analyzed by ASV in the manner described above. Given the sample data in Table 5, calculate the current concentration levels of the heavy metals (Cu, Pb and Zn) in suspended solids in Lake Nakuru. What do the data tell you about the heavy metal concentrations in the suspended solids at Lake Nakuru? Have the concentrations increased or decreased since the 1996 sampling?
Table 5: Sample Data for Copper, Lead and Zinc Determination in Suspended Solid Samples in Lake Nakuru
|
Sample number |
ip (µA) |
|||||
|
Sample |
Sample + 100 µL spike |
|||||
|
Cu |
Pb |
Zn |
Cu |
Pb |
Zn |
|
|
1 |
0.0999 |
0.1511 |
0.1985 |
0.4697 |
0.9513 |
0.4545 |
|
2 |
0.1001 |
0.1623 |
0.1752 |
0.4898 |
0.9253 |
0.4326 |
|
3 |
0.0959 |
0.1506 |
0.1863 |
0.4800 |
0.9222 |
0.4863 |
Note: All samples analyzed were a 10.00 mL aliquot. The 100.0 µL standard spike contained a target concentration of 5.000 ppm of each metal of interest. The exact concentration (depending on standard mass weighed out) can be calculated.
Chromium measurement
You may have noticed that the data analyzed so far was for lead, copper and zinc, while the Nelson 1998 paper has reported data for chromium. If we wish to use voltammetry for chromium measurement, the technique of choice is Adsorptive Stripping Voltammetry. The key difference between Adsorptive and Anodic stripping voltammetry is in the deposition (accumulation) procedure. In this alternative technique, the accumulation step is purely adsorptive (i.e. the analyte “sticks” onto the electrode) rather than electrolytic (electrochemical deposition). The stripping step for the adsorptive method can involve either anodic or cathodic removal of the analyte from the electrode.
Q15. Let’s see if you are paying attention . . . During the accumulation step in adsorptive stripping voltammetry, does a redox reaction occur?
Note also that the Nelson 1998 paper measured chromium by graphite furnace atomic absorption spectroscopy. In natural samples, chromium can exist in the Cr(III) oxidation state or in various compounds in the Cr(VI) oxidation state. Graphite furnace AAS is an excellent choice for a total chromium measurement. As previously mentioned, one advantage of electrochemical over spectroscopic methods is the speciation capabilities. While adsorptive stripping methods have been developed to differentiate between Cr(III) and Cr(VI) species (Dominguez 2002), the data below will reflect a total chromium measurement. This analysis will keep this introductory material simpler and allow comparison with the graphite furnace AAS data in Nelson 1998.
In a typical experiment for chromium measurement a HMDE (Golimowski 1985) or mercury-based electrode (Bas 2006 and Wang 1997) is employed as the working electrode. A complexing agent is added to complex with chromium(III), forming a surface-active complex that adsorbs to the electrode surface. During this deposition step, the electrode is held at a potential sufficiently negative to reduce any Cr(VI) to Cr(III). The Cr(III) complex adsorbs to the electrode surface. Subsequently, during the stripping step, the electrode is cathodically scanned and the complex is reduced, giving rise to the analytical signal. The reduction is a one electron process:
\[\ce{Cr(III)} + e^- \rightarrow \ce{Cr(II)}\nonumber\]
Differential Pulse Adsorptive Stripping Voltametry measurements of chromium. The instrumental experimental conditions used are shown in the table below.
Table 6: Experimental conditions for the determination of chromium in sediments*
| Ea | Accumulation potential (mV/Ag, AgCl, KCl satd.) | -1000 |
| Ei | Initial scan potential mV/Ag, AgCl, KCl satd.) | -1000 |
| Ef | Final potential (mV/Ag, AgCl, KCl satd.) | -1600 |
| dE/dt | Potential scan rate (mV/s) | 10 |
| f | Pulse repetition (s) | 0.1 |
| ΔE | Pulse amplitude (mV) | 50 |
| v | Pulse duration (ms) | 20 |
| ts | Sampling time (ms) | 8 |
| ta | accumulation time (s) | 180 |
| tr | Rest time (s) | 15 |
| t | Purging time (s) | 300 |
| u | Stirrer speed (r.p.m.) | 600 |
*Note: conditions modified from Wang 1997 and Golimowski 1985.
Simulated sample data acquired for Lake Nakuru sediment samples are given in Table 7. Sample preparation was similar to that described by Locatelli 1999. Briefly, 1 g sediment samples were acid digested and diluted to 100 mL. This solution was diluted by a factor of 20 with appropriate electrolyte. The analysis was performed on this second solution. The single standard addition method of quantitation was used (section 5.3.3 in Analytical Chemistry 2.0). A 10.00 mL aliquot of sample solution was pipetted into the sample container and purged with nitrogen as described above. The sample underwent the adsorptive stripping procedure outlined above. Upon collection of this voltammogram, 100.0 µL of the standard solution (5.000 ppm) was added to the 10.00 mL sample and the adsorptive stripping procedure was performed on this “spiked” sample. The data for the peak current was recorded in tabular format and is shown below. Use this data to calculate the concentration of each heavy metal in the sediment samples in µg/g (as was reported in Nelson 1998). Don’t forget to account for any dilutions performed during the sample preparation. Hand-calculate the concentration for sample 1 then set up a spreadsheet to calculate the concentrations of the metals for all three samples.
Q16. Recently, sediment and suspended solid samples were collected from Lake Nakuru and analyzed by ASV in the manner described above. Given the sample data in Tables 7 and 8, calculate the current concentration levels of chromium in the sediment samples obtained from Lake Nakuru. What do the data tell you about chromium concentration in the sediments and suspended solids in Lake Nakuru? Has the concentration increased or decreased since the 1996 sampling?
Table 7: Sample Data for Chromium Determination in Sediment Samples in Lake Nakuru
|
Sample number |
ip (µA) |
|
|
Sample |
Sample + 100 µL spike |
|
|
Cr |
Cr |
|
|
1 |
0.1111 |
0.2522 |
|
2 |
0.1212 |
0.2855 |
|
3 |
0.1359 |
0.2989 |
Note: All samples analyzed were a 10.00 mL aliquot. The 100.0 µL standard spike contained a target concentration of 5.000 ppm of Cr(III). The exact concentration (depending on standard mass weighed out) can be determined on the corresponding Excel spreadsheet.
Table 8: Sample Data for Chromium Determination in Suspended Solid Samples in Lake Nakuru
|
Sample number |
ip (µA) |
|
|
Sample |
Sample + 100 µL spike |
|
|
Cr |
Cr |
|
|
1 |
0.1015 |
0.8986 |
|
2 |
0.1985 |
0.9563 |
|
3 |
0.1574 |
0.9254 |
Note: All samples analyzed were a 10.00 mL aliquot. The 100.0 µL standard spike contained a target concentration of 5.000 ppm of Cr(III). The exact concentration (depending on standard mass weighed out) can be determined on the corresponding Excel spreadsheet.
Q17. If your sample was known to contain metal ions that cannot be measured by ASV, what other techniques would you suggest trying?
References and Further Reading
Golimowski, J.; Valenta, P.; Nurnberg, H.W. “Trace Determination of Chromium in Various Water Types by Adsorption Differential Pulse Voltammetry” Fresnius’ J. Anal. Chem., 1985, 322, 315-322.
Locatelli, C.; Astara, A.; Vasca, E.; and Campanella, V. “Voltammetric and spectroscopic determination of toxic metals in sediments and sea water of salerno gulf” Environmental Monitoring and Assessment, 1999, 58, 23-37.
Wang, J. “Chapter 3: Controlled Potential Techniques” in Analytical Electrochemistry, 3rd Ed. 2006, John Wiley and Sons, Inc.: Hoboken, New Jersey.